All tools
Find tools by scientific task, accepted input, application, and run mode. Learn more
AbLang
Restore missing antibody residues, generate 768-dimensional sequence or residue representations, and calculate amino-acid likelihood scores with the original AbLang heavy- and light-chain models.
AbLang-2
Antibody-specific language model for predicting non-germline residues (NGL) in antibody sequences. AbLang-2 addresses germline bias in existing antibody language models by focusing on somatic hypermutation patterns, enabling more accurate prediction of amino acid likelihoods and generation of context-aware embeddings for antibody sequences.
ABodyBuilder3
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
ADMET-AI
Predict ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties from SMILES strings using machine learning models trained on Therapeutics Data Commons datasets.
Admetica
Predict 22 ADMET properties from SMILES strings with the native Admetica Chemprop models from Datagrok.
AF-Cluster
Cluster protein multiple sequence alignments with the source AF-Cluster DBSCAN workflow to prepare subgroup MSAs for alternative-conformation prediction.
AF2BIND
AF2BIND predicts ligand-binding residues from a protein structure using AlphaFold2 pair representations and a 20-residue bait sequence.
AF2Dock
AF2Dock adapts AlphaFold2-style co-folding for structure-based protein-protein docking. It docks receptor and ligand protein structures with flow-matching refinement and ranks sampled complexes by iPTM.
Aggrescan3D
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
AIMNet2
Predict molecular energies, atomic forces, atomic and spin charges, dipole vectors, stress tensors, and Hessians from coordinate-bearing molecular structures with AIMNet2 neural network potentials.
Aliphatic Index
Calculate the aliphatic index of protein sequences. A measure of the relative volume occupied by aliphatic side chains, indicating thermostability.
AllMetal3D
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
AlphaFlow
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.

AlphaFold Database Download
Download AlphaFold DB predicted structures and confidence files by UniProt accession.
AlphaFold2
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.

AlphaGenome
AlphaGenome predicts variant effects on gene expression by comparing reference and alternate alleles. ProteinIQ currently supports RNA-seq analysis windows up to 512K base pairs. Requires a DeepMind API key; ProteinIQ credits scale with window size.
Amino acid composition
Analyze amino acid composition of protein sequences. The tool accepts FASTA sequences and outputs the percentage of each amino acid in the sequence.
ANARCI
Number antibody and T cell receptor variable domain sequences using IMGT, Chothia, Kabat, Martin, AHo, or Wolfguy. Reports chain type, the best HMM species match, and optional germline assignments.
AntiFold
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
AutoDock Vina
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
AutoDock-GPU
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
BindCraft
Design de novo protein binders using AlphaFold2 backpropagation, ProteinMPNN sequence optimization, and PyRosetta relaxation. BindCraft generates novel protein sequences that bind to user-specified target surfaces.
BindingDB Download
Download BindingDB binding record files by BindingDB monomer ID.
BioPhi
Antibody humanization and humanness evaluation platform from Merck. Sapiens mode uses deep learning trained on the Observed Antibody Space (OAS) to humanize antibody sequences, while OASis mode evaluates humanness using 9-mer peptide search against human antibody databases.
Boltz-2
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
BoltzGen
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
Brenk filter
Identify toxic, reactive, and pharmacokinetically problematic molecular fragments using structural alert patterns
CANYA
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
Carbon
Carbon is a DNA language model for generation, scoring, and sequence comparison using the native Hugging Face Carbon model family.
Chai-1
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ChEBI Download
Download ChEBI chemical entity records as JSON by ChEBI ID.
ChEMBL Download
Download ChEMBL molecule records as JSON and SDF by ChEMBL compound ID.
Chou-Fasman
Predict protein secondary structure using the classic Chou-Fasman algorithm based on amino acid propensities
CleaveNet
Official CleaveNet tool for matrix metalloproteinase cleavage prediction and peptide generation. Predict cleavage z-scores plus uncertainty across 18 MMP variants, evaluate against truth z-scores, or generate candidate peptides unconditionally or from MMP z-score profiles.
Clustal Omega
Perform multiple sequence alignment on protein or nucleotide sequences using the Clustal Omega algorithm.
ColabDock
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.
CpG Island Finder
Identify CpG islands in DNA sequences using the Gardiner-Garden and Frommer criteria. Analyze GC content, CpG density, and observed/expected ratios.
CSV to FASTA
Convert CSV and TSV files containing sequence data to FASTA format with flexible column mapping and automatic delimiter detection
DeepEMhancer
DeepEMhancer is a deep learning-based post-processing tool for cryo-EM maps. It performs automatic sharpening, masking, and denoising in a single step without requiring an atomic model. Supports half-map inputs for improved local mask estimation.
DeepImmuno
Predict peptide immunogenicity with DeepImmuno-CNN from peptide sequences and HLA alleles.
DFMDock
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
DiffAb
AI-powered antibody CDR design using equivariant diffusion models. Generates complementarity-determining region (CDR) sequences and structures for antibody structures and antibody-antigen complexes. Supports single- and multi-CDR co-design, antibody optimization, fixed-backbone sequence design, and structure prediction.
DiffDock-L
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DLKcat
DLKcat predicts enzyme turnover numbers (kcat values) from protein sequences and substrate structures using its published deep-learning model.
DNA mutation generator
Mutate DNA online and generate reproducible substitution, insertion, deletion, or mixed variant libraries from plain sequences or FASTA.
DNA Shuffle
Shuffle DNA sequences while preserving nucleotide, dinucleotide, or k-mer composition for generating randomized control sequences
DNA to Protein Converter
Translate DNA sequences to protein sequences using genetic code
DNA to RNA converter
Convert DNA sequences to RNA (transcription) - replaces T with U
DNAGenIQ - Random DNA sequence generator
Generate random DNA sequences with customizable length, GC content, and restriction sites for molecular cloning and testing purposes.
DockQ
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
DR-BERT
DR-BERT is a compact protein language model that predicts intrinsically disordered regions (IDRs) in proteins. It outputs per-residue disorder probability scores (0–1) from amino acid sequences, enabling fast and accurate annotation of disordered regions without structural data.
DSSP
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.
DynamicBind
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
EquiDock
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
ESM-2
ESM-2 is a 650M parameter protein language model from Meta AI trained on 250M protein sequences. Generate rich sequence representations for downstream tasks like structure prediction, function annotation, and variant effect prediction.
ESM-C
ESM-C generates protein sequence representations and optional forward-pass sequence logits using Biohub protein language models. It supports the 300M, 600M, and 6B model variants for embedding extraction from tokenizer-compatible protein sequences.
ESM-IF1
Inverse folding with ESM-IF1. Design protein sequences for given 3D backbone structures using a geometric deep learning model. Generate multiple sequence variants optimized for your target structure.
ESMfold
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ESMFold2
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
eToxPred
Predict toxicity and synthetic accessibility of small molecules using machine learning. eToxPred combines toxicity risk assessment with synthetic accessibility scoring to help prioritize drug candidates.
EvoDiff
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
EvoIF
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
EvoPro
Optimize protein binders using genetic algorithms combined with AlphaFold2 fitness evaluation and ProteinMPNN sequence design. EvoPro evolves protein sequences to maximize binding affinity and structural quality through iterative cycles of mutation, selection, and validation.
Extinction coefficient calculator
Calculate the molar extinction coefficient of protein sequences at 280 nm. Used for protein concentration determination by UV spectroscopy.
FASTA splitter
Split FASTA files into record-based chunks by sequence count, target file count, maximum residues per file, or one file per sequence.
FASTA to FASTQ Converter
Convert FASTA sequence files to FASTQ format with mock quality scores
FASTQ to FASTA converter
Convert standard FASTQ reads to FASTA with validation, IUPAC nucleotide support, average-quality filtering, and downloadable summaries
FastTree
Infer approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences.
Filter DNA
Clean and filter DNA sequences by removing or replacing non-standard nucleotide characters. Supports multiple filter modes including standard 4 bases, IUPAC ambiguity codes, and custom character sets.
Filter protein
Clean and filter protein sequences by removing or replacing non-standard amino acid characters. Supports multiple filter modes including standard 20 amino acids, IUPAC codes, and custom character sets.
FindPept
Match experimental peptide masses against theoretical digest fragments of a protein sequence. Identify peptides from mass spectrometry data by peptide mass fingerprinting.
FlowDock
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
FoldSeek
Fast protein structure search, comparison, and clustering. Search your structure against 200M+ AlphaFold predictions, compare 2 structures, or cluster up to 2500.
fpocket
Open-source protein pocket detection using Voronoi tessellation and alpha spheres. Identifies ligand binding sites with druggability scores.
GC content calculator
Calculate GC content, GC/AT skew, melting temperature, and CpG islands for DNA/RNA sequences, with a sliding-window GC plot. Analyze individual sequences or get combined statistics.
GenBank Feature Extractor
Extract sequence features (CDS, mRNA, gene, etc.) from GenBank files in FASTA format with support for spliced features
GenBank to FASTA Converter
Convert GenBank files to FASTA format
Genie 3
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
GenMol
GenMol is a generative AI model from NVIDIA that creates novel drug-like molecules using masked discrete diffusion. It generates molecules in SAFE representation format and supports de novo generation, linker design, motif extension, and scaffold decoration.
GeoDock
GeoDock predicts flexible protein-protein docking complexes from two separate protein structures using a multi-track iterative transformer and the DIPS 0.3 checkpoint from the Gray Lab release.
Glycosylation site finder
Find potential N-linked glycosylation sites (NX[S/T] sequons) in protein sequences. Identifies asparagine residues in the consensus motif for N-glycosylation.
gmx_MMPBSA
Calculate binding free energies using MM/PBSA and MM/GBSA methods for protein-ligand, protein-protein, and protein-DNA complexes. Provides detailed energy decomposition and per-residue contributions.
GNINA
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
GRAVY
Calculate the GRAVY (Grand Average of Hydropathy) score of protein sequences. Positive values indicate hydrophobic proteins, negative values indicate hydrophilic proteins.
GROMACS
Run molecular dynamics simulations using the GROMACS engine with classical force fields (AMBER, CHARMM, GROMOS, OPLS). Study protein dynamics, conformational flexibility, and structural stability with production-grade MD methodology.
HADDOCK3
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
HighFold
Cyclic peptide structure prediction using HighFold, a modified ColabFold/AlphaFold2 framework with CycPOEM (Cyclic Position Offset Encoding Matrix) for head-to-tail and disulfide bridge constraints.
HMMER
Sensitive sequence homology search using profile hidden Markov models. More accurate than BLAST for detecting remote homologs, ideal for finding evolutionarily distant protein family members.
Humatch
Humatch is an antibody humanization tool that transforms non-human antibody sequences into humanized variants. Uses three lightweight CNNs to identify optimal human V-genes and generate paired heavy and light chain sequences with minimal edits while maintaining functionality.
Hydropathy plot
Generate Kyte-Doolittle hydropathy plots to visualize hydrophobic and hydrophilic regions along protein sequences. Identify transmembrane domains and surface-exposed regions.
Hydrophobicity plot
Generate hydrophobicity plots using 24 different amino acid scales. Visualize hydrophobic and hydrophilic regions for protein analysis, epitope prediction, and membrane protein studies.
HyperMPNN
Design thermostable protein sequences using ProteinMPNN trained on hyperthermophilic organism structures. Generates sequences optimized for improved thermal stability without requiring ligands or additional context.
IgBLAST
Analyze immunoglobulin (antibody) and T cell receptor variable domain sequences. Identifies V/D/J gene segments, delineates CDR regions, and analyzes rearrangement junctions.
IgDesign
Design antibody heavy- and light-chain CDR sequences from an antibody-antigen complex with the IgDesign inverse-folding model.
IgGM
IgGM is a generative foundation model for antibody and nanobody design against a target antigen. Supports CDR design, affinity maturation, inverse design, and framework design. Requires an antigen structure (PDB) and antibody sequences with "X" marking positions to design.
ImmuneBuilder
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
InChI to SMILES
Convert InChI strings into SMILES with batch support and downloadable outputs.
Instability Index
Calculate the instability index of protein sequences. Values above 40 indicate an unstable protein with a short half-life in vitro.
IntelliFold 2
Controllable all-atom structure prediction for proteins, ligands, DNA, RNA, and multi-component complexes using IntelliFold 2.0.4 on its AlphaFold 3 JAX engine.
InterPro Download
Download InterPro protein family and domain records as JSON by InterPro ID.
IPC 2.0 (isoelectric point calculator)
Isoelectric Point Calculator 2.0 - Predict protein/peptide isoelectric point (pI) using 18+ validated pKa scales, SVR models, and deep learning. Supports proteins, peptides, and comprehensive analysis.
IPSAE
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
IQ-TREE
Build phylogenetic trees using maximum likelihood with automatic model selection (ModelFinder) and ultrafast bootstrap support.
Lead-likeness filter
Screen for lead-like compounds using stricter molecular descriptor criteria than Lipinski or Veber rules for early-stage drug discovery
Ligand fixer
Fix ligand files that fail RDKit, Meeko, or docking preparation. Repair SDF, MOL, and MOL2 inputs, apply safe chemistry cleanup, and export docking-ready SDF files.
LigandMPNN
Design protein sequences with atomic context from ligands, metals, and nucleotides. Achieves 63.3% sequence recovery at binding sites, significantly outperforming ProteinMPNN (50.5%).
LightDock
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
Lipinski's rule of 5
Lipinski's Rule of Five predicts whether compounds will be orally bioavailable by evaluating molecular weight, LogP, hydrogen bond donors, and acceptors.
LMI4Boltz
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
LocScale
LocScale performs physics-informed local sharpening of cryo-EM density maps using half-maps or full MRC/MAP volumes, with optional mask and reference-map inputs.
MAFFT
Perform multiple sequence alignment using MAFFT (Multiple Alignment using Fast Fourier Transform). Supports multiple algorithms from fast progressive to highly accurate iterative methods.
mBER
Design VHH nanobody binders using AlphaFold-Multimer with structure templates and sequence conditioning. mBER (Manifold Binder Engineering and Refinement) generates novel VHH antibody sequences that bind to user-specified target proteins.
MD Trajectory Analysis
Analyze molecular dynamics trajectories using a ProteinIQ tool pinned to MDAnalysis 2.9.0. Calculate RMSD, residue-aggregated RMSF, radius of gyration, distance tracking, and additional trajectory observables from standard topology and trajectory files.
MDGen
MDGen is a generative AI model for molecular dynamics trajectory generation. Generate physically plausible conformational ensembles from a single protein structure, enabling rapid exploration of protein dynamics without expensive MD simulations.
MiniFold
MiniFold is a fast single-sequence protein structure predictor that is 10-20x faster than ESMFold. It predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it ideal for rapid structure prediction.
MMseqs2
Ultra-fast sequence search and clustering. 10,000x faster than BLAST for database searches, with powerful sequence clustering capabilities for proteins and nucleotides.
MOL to SMILES
Convert MDL MOL ligand files into canonical SMILES strings for registration, filtering, and downstream analysis.
MOL2 to SMILES
Convert MOL2 ligand files into SMILES strings for registration, filtering, and downstream analysis.
Molecular descriptors
Compute 200+ RDKit molecular descriptors, drug-likeness rule violations, and structural fingerprints for QSAR, virtual screening, and ML workflows
MolProbity
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
MUMmer4
Rapidly align and compare DNA sequences using MUMmer4 nucmer. Perform pairwise genome comparisons to identify SNPs, indels, and structural variants between reference and query genomes.
MUSCLE5
Align protein, DNA, or RNA sequences with MUSCLE v5.3 using the native PPP or Super5 algorithm, with optional stratified and diversified ensembles.
NetSolP-1.0
Predict protein solubility and usability for E. coli expression using ESM protein language models
ODesign
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
Oligo analyzer
Calculate DNA oligo melting temperature, molecular weight, extinction coefficient, GC content, and screen for hairpins, self-dimers, and primer-pair dimers.
One-to-Three Converter
Convert single-letter amino acid codes to three-letter codes
Open Babel
Run Open Babel in the browser to convert chemical and structure files, with coordinate, hydrogen, and pH options where the WASM runtime supports them.
OpenFE
Calculate absolute hydration free energies (AHFE) for neutral small molecules with OpenFE and GPU-accelerated OpenMM simulations.
OpenFold-3
OpenFold-3 is an open-source AI model for biomolecular structure prediction, aiming to reproduce AlphaFold3. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
OpenMM
Run GPU-accelerated molecular dynamics simulations using OpenMM. Simulate protein and protein-ligand complex dynamics with industry-standard force fields (AMBER, CHARMM) and OpenFF ligand parameterization.
ORB v3
ORB v3 is a universal interatomic potential (machine learning force field) that predicts energies, forces, and stress tensors for atomic systems. Supports both molecular and materials structures with geometry optimization using conservative and direct model variants.
ORF Finder
Find open reading frames in DNA with the native NCBI ORFfinder search, translations, coordinates, and output files.
PAINS filter
Screen compounds for Pan-Assay Interference patterns that cause false positives in biological assays
PandaDock
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
ParaSurf
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
PDB Download
Download PDB, CIF, and FASTA files from RCSB PDB by 4-character structure ID.
PDB to CIF Converter
Convert Protein Data Bank files to Crystallographic Information File format

PDB to FASTA converter
Convert Protein Data Bank files to FASTA sequence format
PDB to MOL2 Converter
Convert Protein Data Bank files to MOL2 molecular format
PDB to SDF Converter
Convert Protein Data Bank files to Structure Data Format
PDB2PQR
PDB2PQR prepares protein structures for electrostatics calculations by adding missing atoms, predicting protonation states using PROPKA, and assigning atomic charges and radii from standard force fields.
PDBe Download
Download PDBe structure files as PDB and CIF by PDB ID.
PDBFixer
PDBFixer is an OpenMM-based tool used for fixing problems in protein/DNA/RNA structure files, including adding missing atoms, adding missing residues, and fixing improper formatting.
PDBsum
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
PepMimic
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
PepMLM
Design linear peptide binders for target proteins using a target sequence-conditioned masked language model. PepMLM generates peptide sequences optimized to bind specific protein targets based on ESM-2 protein language modeling.
Peptide cutter
Predict protease and chemical cleavage sites across a protein sequence for up to 39 enzymes simultaneously. Identify where each enzyme cuts, the cleavage residue, and context window around each site.
Peptide mass calculator
Cleave a protein sequence with a chosen protease and compute the masses of the resulting peptides. Supports multiple enzymes, missed cleavages, chemical modifications, and different ion types for mass spectrometry experiment planning.

PeptideBuilder
Build all-atom peptide PDB structures from amino acid sequences using PeptideBuilder geometry defaults, with optional backbone angle controls for simple model peptides.
pI Calculator
Calculate the theoretical isoelectric point (pI) of protein sequences. The pI is the pH at which a protein carries no net electrical charge.
PLIP
Analyze noncovalent interactions in protein-ligand complex structures with PLIP, including hydrogen bonds, hydrophobic contacts, pi interactions, salt bridges, water bridges, halogen bonds, and metal complexes.
PocketFlow
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
PocketXMol
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
PoseBusters
PoseBusters validates generated or docked molecular poses with chemically and structurally grounded quality checks for molecular geometry, intermolecular interactions, and optional reference-pose agreement.
PPAP
PPAP predicts protein-protein binding affinity as −ΔG and Kd from a multi-chain PDB complex using interfacial graph features and ESM2-3B embeddings.
Primer3
Design PCR primers for DNA sequences with scientifically faithful Primer3 controls for target regions, thermodynamics, product size, and primer quality.
ProFam
ProFam-1 is a protein family language model for family-conditioned sequence generation. Provide a protein family in FASTA, A2M, or A3M format and generate new sequences with model likelihood scores for downstream ranking and screening.
ProGen2
ProGen2 is Salesforce Research's protein language model suite for prompt-based de novo protein sequence generation and bidirectional sequence likelihood scoring.
ProLIF
ProLIF calculates protein-ligand interaction fingerprints from 3D structures, returning residue-level interaction tables, interaction metadata, and native fingerprint files.
PROPKA 3
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
ProstT5
ProstT5 is a protein language model that bidirectionally translates between amino acid sequences and 3Di structural tokens. It enables fast structure-based searches and inverse folding by encoding structural information into a sequence-like representation.
Prot2Prop
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.
Protein charge plot
Plot net charge vs pH for protein sequences. Visualize how protein charge changes across pH 0-14 and identify the isoelectric point (pI) where the net charge crosses zero.
Protein molecular weight calculator
Calculate protein molecular weight (MW) from amino acid sequences in Daltons and kilodaltons. Supports FASTA and CSV sequence input, average or monoisotopic masses, initiator Met removal, and disulfide correction.
Protein motif scanner
Scan protein sequences for biologically important motifs including glycosylation sites, phosphorylation sites, nuclear localization signals, prenylation motifs, and more.
Protein parameters
Calculate protein parameters, including molecular weight, theoretical pI, extinction coefficients, aromaticity, secondary structure fractions, atomic composition, estimated half-life, and several indices, including instability, aliphatic index, and GRAVY.
Protein scale profiler
Generate amino acid property profiles using 42 different scales spanning hydrophobicity, secondary structure propensity, flexibility, polarity, surface accessibility, antigenicity, and more.
Protein stability
Predict protein stability using validated BioPython methods: Instability Index, Aliphatic Index, GRAVY, flexibility analysis, and charge distribution
Protein to DNA converter
Reverse translate protein sequences to possible DNA sequences
Protein-Sol
Predict protein solubility from amino acid sequence using the University of Manchester Protein-Sol method.
Proteina-Complexa
Design protein binders against a target structure with NVIDIA BioNeMo's Proteina-Complexa generative pipeline.
ProteinMPNN
Design protein sequences for given backbone structures using deep learning. Fast and accurate inverse folding with state-of-the-art sequence recovery (52.4%).
Protenix v2
Enhanced Protenix v2 biomolecular structure prediction by ByteDance. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Proteo-R1
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
ProtGenIQ - Random protein sequence generator
Generate random protein sequences with customizable length, composition, and amino acid properties
PubChem Download
Download PubChem compound records as JSON, SDF, and SMILES by CID, compound name, or InChIKey.
pyRMSD
Calculate pairwise RMSD matrices for PDB structure ensembles with pyRMSD, including the condensed matrix and source statistics files.
pySCA
Statistical Coupling Analysis for protein families. Identifies co-evolving residue groups (sectors) from multiple sequence alignments using the SCA method from the Ranganathan Lab.
QEPPI
Quantitative estimate for protein-protein interaction inhibitor potential. Evaluates drug-likeness for compounds targeting PPIs.
QuickGO Download
Download QuickGO Gene Ontology term records as JSON by GO ID.
Radius of gyration
Calculate the radius of gyration (Rg) for protein structures from PDB files. Supports multiple chains and atom selection options.
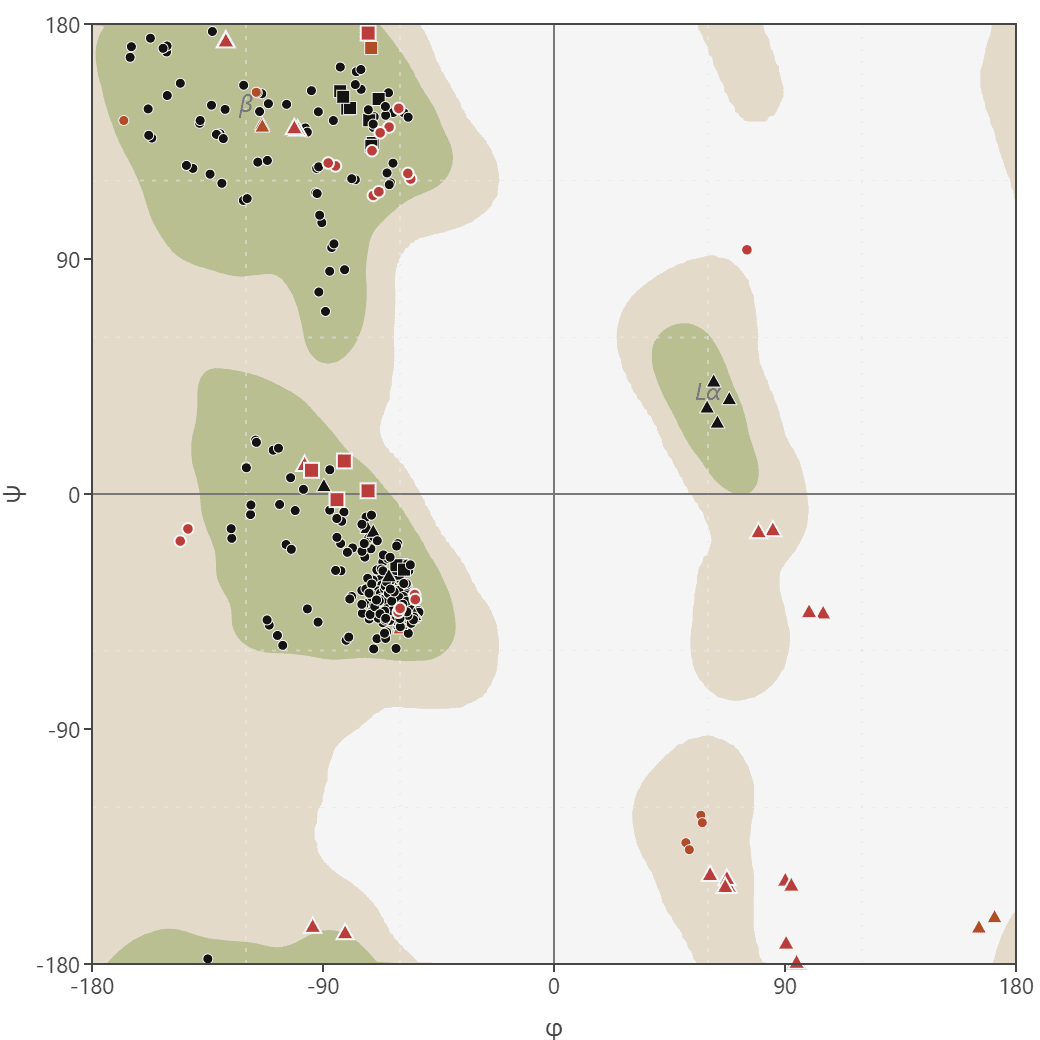
Ramachandran plot
Generate Ramachandran plots from PDB or mmCIF structures to analyze protein backbone dihedral angles (phi/psi). Visualize favored, allowed, and outlier regions.
RAxML-NG
Perform maximum-likelihood phylogenetic tree inference with RAxML-NG for aligned protein or DNA sequences. Supports ML search, bootstrap analysis, and native automatic model-family selection.
Reverse complement generator
Generate reverse, complement, or reverse-complement of DNA/RNA sequences
RFantibody
Structure-based de novo antibody and nanobody design pipeline combining antibody-tuned RFdiffusion, ProteinMPNN sequence design, and antibody-tuned RoseTTAFold2 filtering.
RFdiffusion
RFdiffusion is a state-of-the-art protein structure generation tool that uses diffusion models to design proteins de novo, create binders, scaffold motifs, and generate symmetric oligomers with atomic precision.
RFdiffusion 2
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
RFdiffusion3
All-atom generative diffusion model for protein design with complex constraints. Design binders, enzymes, and symmetric protein assemblies.
Rhea Download
Download Rhea biochemical reaction records as RDF by Rhea reaction ID.
RMSD calculator
Calculate RMSD between protein structures with optimal rigid-body alignment, explicit atom correspondence, coverage diagnostics, per-residue results, and fitted coordinate files.
RNA to DNA converter
Convert RNA sequences to DNA (reverse transcription) - replaces U with T
RNAalifold
RNAalifold computes consensus RNA secondary structure from a multiple sequence alignment. Uses covariation information to improve prediction accuracy for evolutionarily conserved structures.
RNAcofold
RNAcofold predicts the joint secondary structure of two interacting RNA molecules and optionally reports partition-function and concentration-dependent equilibrium metrics.
RNAdistance
RNAdistance compares RNA secondary structures using the selected native ViennaRNA distance representation and comparison mode.
RNAdos
RNAdos calculates density-of-states summaries for RNA sequences, reporting representative structures and state counts across energy bands.
RNAduplex
RNAduplex computes the hybridization structure between two RNA sequences. Predicts the optimal duplex formation and binding energy.
RNAeval
RNAeval calculates the free energy of an RNA secondary structure for a given sequence. Evaluates if a proposed structure is thermodynamically favorable.
RNAfold
RNAfold predicts RNA secondary structure using minimum free energy (MFE) algorithms and optionally returns partition-function ensemble metrics when explicitly enabled.
RNAGenIQ - Random RNA sequence generator
Generate random RNA sequences with customizable types and structural features
RNAinverse
RNAinverse designs RNA sequences for a specified target secondary structure using ViennaRNA inverse-folding semantics.
RNALfold
RNALfold reports locally stable RNA secondary structures within a sliding window and returns their start and end positions on the input sequence.
RNAplex
RNAplex predicts fast query-target RNA interactions, reporting parsed hit coordinates, structures, and energies.
RNAplfold
RNAplfold computes local base pair probabilities using a sliding window approach. Useful for analyzing accessibility and identifying binding sites in long RNA sequences.
RNAplot
RNAplot renders ViennaRNA secondary-structure plot files from a supplied RNA sequence and dot-bracket structure.
RNAsubopt
RNAsubopt enumerates all RNA secondary structures within a specified energy range above the minimum free energy (MFE). Useful for exploring the structural ensemble and identifying alternative conformations.
RNAup
RNAup predicts accessibility-aware RNA-RNA interactions, reporting opening-energy terms alongside interaction energies and downloadable native output files.
RosettaFold3
Open-source structure prediction neural network for proteins, nucleic acids, and small molecules. State-of-the-art accuracy with multi-chain support.
Salmon
Quantify transcript abundance from RNA-seq reads with Salmon selective alignment. Upload a transcript FASTA reference plus single-end or paired-end FASTA/FASTQ reads to produce TPM and estimated read-count tables.
SASA calculator
Calculate Solvent Accessible Surface Area (SASA) for protein structures using the Shrake-Rupley algorithm.
ScanNet
Geometric deep learning model for predicting protein binding sites directly from 3D structure. Identifies where proteins interact with other proteins, antibodies, or disordered proteins with high accuracy, including for novel protein folds.
SDF to PDB Converter
Convert Structure Data Format files to Protein Data Bank format
SDF to SMILES
Convert SDF ligand files, including multi-record batches, into SMILES strings.
SigmaDock
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
SMILES to InChI
Convert SMILES strings into InChI strings with batch support and downloadable outputs.
SMILES to MOL2
Convert SMILES strings into 3D MOL2 files for docking and molecular modeling workflows.
SMILES to PDB
Convert SMILES strings into 3D PDB files for molecular visualization and downstream docking preparation.
SMILES to SDF
Convert single SMILES strings or small batches into 3D SDF files with downloadable per-entry files and a combined batch output.
SMINA
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
SMRTnet
Deep learning framework for predicting small molecule-RNA interactions using RNA secondary structure. Combines language models, CNNs, and graph attention networks for binding prediction.
SolubleMPNN
Specialized model for soluble protein sequence design. Trained exclusively on soluble proteins for optimized performance on cytoplasmic and extracellular proteins.
SPRINT
Rank a compound library against one protein target with SPRINT protein and ligand co-embeddings and native cosine similarity.
StringZilla v5
Calculate batched Levenshtein distances and Needleman–Wunsch or Smith–Waterman sequence scores with StringZilla v5 CPU acceleration.
SuperWater
Predict protein hydration sites from a structure using a diffusion model with ESM features and a confidence-filtering head.
SurfDock
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
TEMPL Pipeline
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
ThermoMPNN
Predict protein thermostability changes (ΔΔG) for point mutations using a graph neural network. Enables computational saturation mutagenesis screening to identify stabilizing mutations.
Three-to-one converter
Convert three-letter amino acid codes to single-letter codes
TLimmuno2
Predict MHC class II peptide immunogenicity (CD4+ T cell response) using transfer learning with LSTM.
ToxPred 2.0 (Toxicity prediction)
Screen compounds for structural toxicity alerts using PAINS, Brenk, and NIH filters. For focused screening, see PAINS Filter, Brenk Filter, or Veber's Rule.

TXT to FASTA converter
Convert TXT or plain text sequences into FASTA format files for DNA, RNA, and protein workflows with cleanup, validation, and downloads
UniProt Download
Download UniProt protein sequence records as FASTA and JSON by accession.
USAlign
USAlign (Universal Structure Alignment) aligns protein, RNA, and DNA structures to compute TM-scores and generate superposed structures. Compare 3D structures to assess structural similarity.
Veber's rule
Veber's Rule predicts oral bioavailability by evaluating molecular weight, LogP, hydrogen bond donors/acceptors, and rotatable bonds
ViennaRNA
ViennaRNA supports a curated set of scientifically faithful ViennaRNA 2.7.2 workflows for RNA folding, density-of-states analysis, interaction prediction, local accessibility, plotting, inverse folding, and structure analysis.