Align protein or nucleotide sequences with selectable accuracy and speed trade-offs. Learn more
Run
5 credits
Output
Configure inputs to begin
Set options on the left, then click “Align Sequences”.
Align protein or nucleotide sequences with selectable accuracy and speed trade-offs. Learn more
Configure inputs to begin
Set options on the left, then click “Align Sequences”.
Align protein or nucleotide sequences with selectable accuracy and speed trade-offs. Learn more
Configure inputs to begin
Set options on the left, then click “Align Sequences”.
MAFFT (Multiple Alignment using Fast Fourier Transform) is a multiple sequence alignment program that aligns protein or nucleotide sequences to identify conserved regions and evolutionary relationships. It offers a range of algorithms that trade off between speed and accuracy, from ultra-fast progressive methods for thousands of sequences to highly accurate iterative methods for smaller datasets.
MAFFT's distinctive feature is its use of Fast Fourier Transform to rapidly identify homologous regions between sequences, dramatically reducing computation time compared to traditional dynamic programming approaches. For alternative alignment approaches, see Clustal Omega which uses HMM-based profile alignment.
MAFFT converts amino acid sequences into numerical representations based on volume and polarity, then uses FFT to rapidly compute correlations between sequences. This identifies conserved regions that serve as anchors, restricting the search space for subsequent dynamic programming alignment.
Traditional sequence comparison requires operations for two sequences of length using dynamic programming. MAFFT reduces this by converting sequences to waves based on physicochemical properties:
Each residue is represented as a 2D vector of volume and polarity values. The correlation between two sequence waves can be computed in time using FFT. Peaks in the correlation identify conserved regions that become anchors for alignment.
MAFFT offers two fundamental strategies:
Progressive methods (FFT-NS-1, FFT-NS-2) build a guide tree from pairwise distances and align sequences following the tree order. FFT-NS-2 improves on FFT-NS-1 by rebuilding the guide tree from the initial alignment before a second pass.
Iterative refinement methods (L-INS-i, G-INS-i, E-INS-i) start with a progressive alignment, then repeatedly refine it. Each iteration divides the alignment into two groups and realigns them, continuing until the score stops improving.
The iterative methods incorporate consistency scores derived from all pairwise alignments. If residues A-B are aligned in sequence pair 1-2, and B-C are aligned in pair 2-3, consistency suggests A-C should align in pair 1-3. This transitive information improves accuracy for divergent sequences.
MAFFT provides multiple algorithms optimized for different scenarios. Choosing the right one depends on your dataset size and accuracy requirements.
Uses local pairwise alignment (Smith-Waterman) with iterative refinement. Best for sequences containing a single alignable domain with flanking regions—the flanking sequences are effectively ignored during alignment.
We recommend L-INS-i for smaller datasets where accuracy is paramount. In Auto mode, MAFFT selects full L-INS-i only when there are fewer than 100 sequences and the longest sequence is under 3,000 residues; larger inputs progressively use lighter strategies according to sequence count and maximum length.
Uses global pairwise alignment (Needleman-Wunsch) with iterative refinement. Assumes the entire sequence length should be aligned, making it suitable for full-length protein domains without terminal extensions.
Use G-INS-i when you know your sequences are globally homologous from end to end.
Uses generalized affine gap costs, allowing long internal gaps without excessive penalty. Designed for sequences with conserved motifs embedded in variable-length regions.
E-INS-i is the most versatile option when you're uncertain about sequence structure. It handles cases where both L-INS-i and G-INS-i might struggle.
A progressive method that builds the guide tree twice—once from 6-mer distances, once from the initial alignment. Much faster than iterative methods with reasonable accuracy.
Use FFT-NS-2 for datasets of 500-10,000 sequences where speed matters more than maximum accuracy.
Uses tree-based clustering for initial distance estimation, enabling alignment of tens of thousands of sequences. Accuracy is lower than other methods but computation remains tractable.
Use PartTree only for datasets exceeding 10,000 sequences where other methods are too slow.
MAFFT auto-detects whether sequences are protein or nucleotide by examining character composition. Manual selection is useful for ambiguous cases like very short sequences.
For nucleotide alignments, MAFFT applies a different scoring matrix optimized for DNA/RNA. The distinction between DNA and RNA affects only the character set validation—alignment scoring is identical.
-. Compatible with FastTree and most downstream tools.The output alignment shows homologous positions as columns. Gap characters (-) indicate insertions or deletions relative to other sequences.
The Files tab includes the alignment and MAFFT's native diagnostic log. The log records the strategy that Auto actually selected, the runtime version, thread count, and any warnings produced during alignment.
Alignment length is the total number of columns, which exceeds any individual sequence length due to gaps. A good alignment minimizes scattered gaps and maximizes contiguous aligned blocks.
Conserved columns (identical residues across all sequences) suggest functional importance. Semi-conserved positions (similar amino acids) often indicate structural constraints.
Both tools produce high-quality alignments, but they excel in different scenarios:
MAFFT's iterative methods (L-INS-i, E-INS-i) generally achieve higher accuracy for difficult alignments with divergent sequences. The FFT-based approach is particularly effective when sequences have variable-length insertions.
Clustal Omega scales better to very large datasets (>10,000 sequences) thanks to its mBed algorithm. Its HMM-based profile alignment can be more robust for sequences with low complexity regions.
For most users with datasets under 1,000 sequences, either tool produces excellent results. We recommend trying both on a subset and comparing the alignments visually.
Multiple sequence alignment is typically the first step in analysis pipelines:
MAFFT assumes input sequences are homologous. It will produce an alignment for any input, but unrelated sequences yield meaningless results. Verify evolutionary relationships before aligning.
The iterative methods (L-INS-i, G-INS-i, E-INS-i) become slow for datasets exceeding a few hundred sequences. Switch to FFT-NS-2 or PartTree for larger datasets.
Very divergent sequences (below ~20% identity for proteins) may not align reliably with any sequence-based method. Consider structure-based alignment with USAlign for such cases.
Align protein, DNA, or RNA sequences with MUSCLE v5.3 using the native PPP or Super5 algorithm, with optional stratified and diversified ensembles.
Calculate batched Levenshtein distances and Needleman–Wunsch or Smith–Waterman sequence scores with StringZilla v5 CPU acceleration.
Ultra-fast sequence search and clustering. 10,000x faster than BLAST for database searches, with powerful sequence clustering capabilities for proteins and nucleotides.
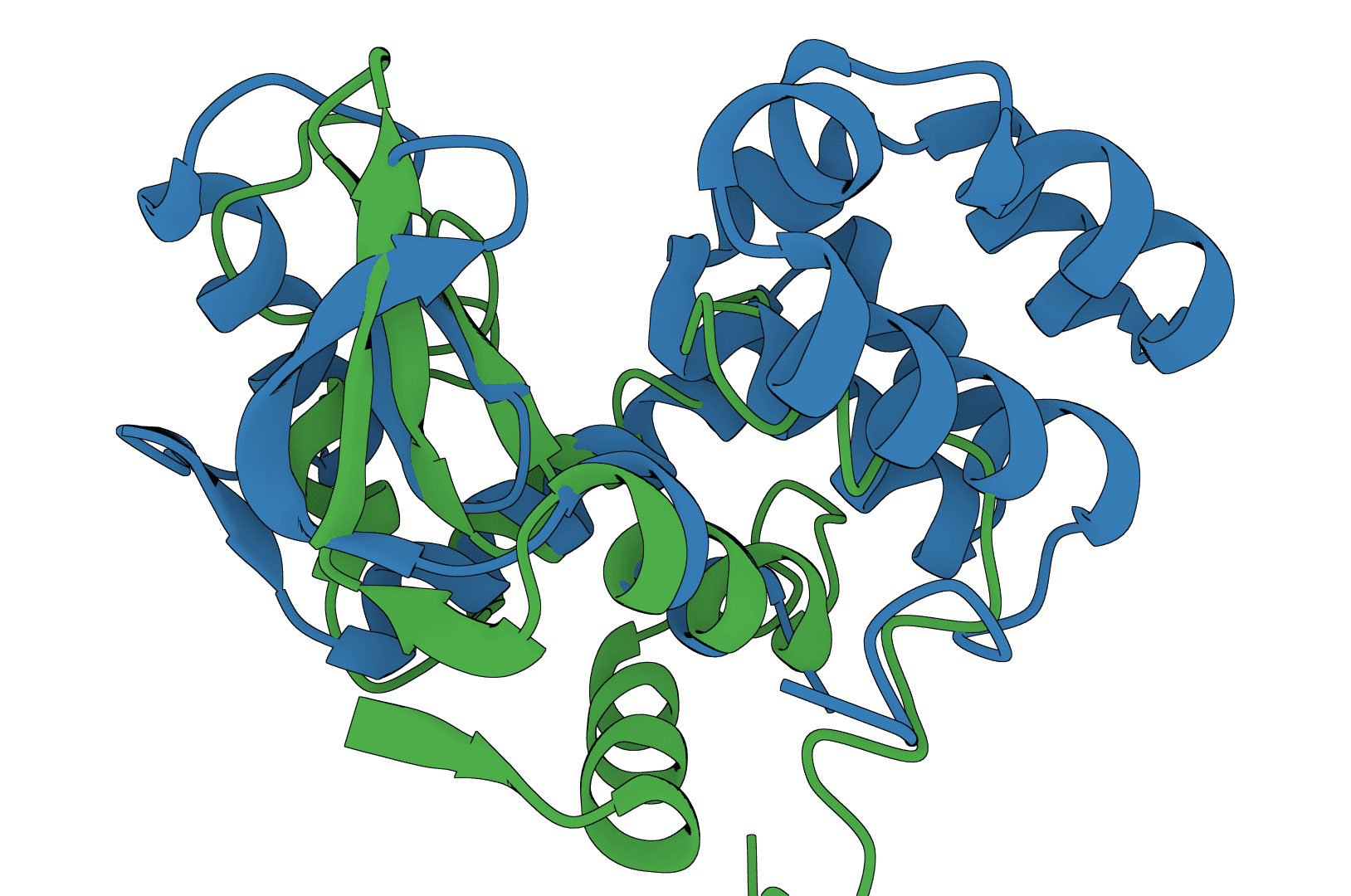
USAlign (Universal Structure Alignment) aligns protein, RNA, and DNA structures to compute TM-scores and generate superposed structures. Compare 3D structures to assess structural similarity.
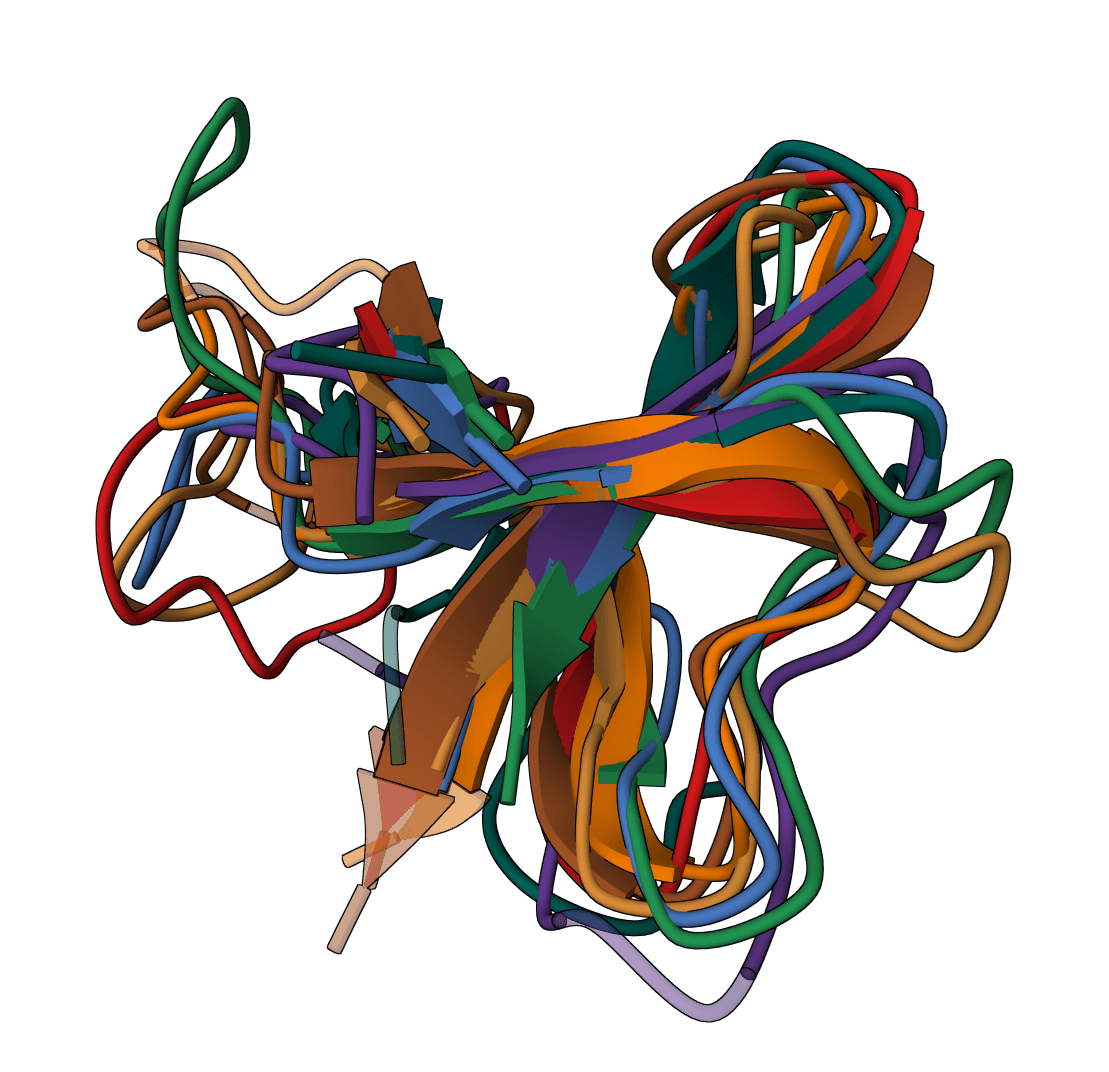
Perform multiple sequence alignment on protein or nucleotide sequences using the Clustal Omega algorithm.
Infer approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences.
Build phylogenetic trees using maximum likelihood with automatic model selection (ModelFinder) and ultrafast bootstrap support.
Rapidly align and compare DNA sequences using MUMmer4 nucmer. Perform pairwise genome comparisons to identify SNPs, indels, and structural variants between reference and query genomes.
Analyze immunoglobulin (antibody) and T cell receptor variable domain sequences. Identifies V/D/J gene segments, delineates CDR regions, and analyzes rearrangement junctions.
Fast protein structure search, comparison, and clustering. Search your structure against 200M+ AlphaFold predictions, compare 2 structures, or cluster up to 2500.
MAFFT (Multiple Alignment using Fast Fourier Transform) is a multiple sequence alignment program that aligns protein or nucleotide sequences to identify conserved regions and evolutionary relationships. It offers a range of algorithms that trade off between speed and accuracy, from ultra-fast progressive methods for thousands of sequences to highly accurate iterative methods for smaller datasets.
MAFFT's distinctive feature is its use of Fast Fourier Transform to rapidly identify homologous regions between sequences, dramatically reducing computation time compared to traditional dynamic programming approaches. For alternative alignment approaches, see Clustal Omega which uses HMM-based profile alignment.
MAFFT converts amino acid sequences into numerical representations based on volume and polarity, then uses FFT to rapidly compute correlations between sequences. This identifies conserved regions that serve as anchors, restricting the search space for subsequent dynamic programming alignment.
Traditional sequence comparison requires operations for two sequences of length using dynamic programming. MAFFT reduces this by converting sequences to waves based on physicochemical properties:
Each residue is represented as a 2D vector of volume and polarity values. The correlation between two sequence waves can be computed in time using FFT. Peaks in the correlation identify conserved regions that become anchors for alignment.
MAFFT offers two fundamental strategies:
Progressive methods (FFT-NS-1, FFT-NS-2) build a guide tree from pairwise distances and align sequences following the tree order. FFT-NS-2 improves on FFT-NS-1 by rebuilding the guide tree from the initial alignment before a second pass.
Iterative refinement methods (L-INS-i, G-INS-i, E-INS-i) start with a progressive alignment, then repeatedly refine it. Each iteration divides the alignment into two groups and realigns them, continuing until the score stops improving.
The iterative methods incorporate consistency scores derived from all pairwise alignments. If residues A-B are aligned in sequence pair 1-2, and B-C are aligned in pair 2-3, consistency suggests A-C should align in pair 1-3. This transitive information improves accuracy for divergent sequences.
MAFFT provides multiple algorithms optimized for different scenarios. Choosing the right one depends on your dataset size and accuracy requirements.
Uses local pairwise alignment (Smith-Waterman) with iterative refinement. Best for sequences containing a single alignable domain with flanking regions—the flanking sequences are effectively ignored during alignment.
We recommend L-INS-i for smaller datasets where accuracy is paramount. In Auto mode, MAFFT selects full L-INS-i only when there are fewer than 100 sequences and the longest sequence is under 3,000 residues; larger inputs progressively use lighter strategies according to sequence count and maximum length.
Uses global pairwise alignment (Needleman-Wunsch) with iterative refinement. Assumes the entire sequence length should be aligned, making it suitable for full-length protein domains without terminal extensions.
Use G-INS-i when you know your sequences are globally homologous from end to end.
Uses generalized affine gap costs, allowing long internal gaps without excessive penalty. Designed for sequences with conserved motifs embedded in variable-length regions.
E-INS-i is the most versatile option when you're uncertain about sequence structure. It handles cases where both L-INS-i and G-INS-i might struggle.
A progressive method that builds the guide tree twice—once from 6-mer distances, once from the initial alignment. Much faster than iterative methods with reasonable accuracy.
Use FFT-NS-2 for datasets of 500-10,000 sequences where speed matters more than maximum accuracy.
Uses tree-based clustering for initial distance estimation, enabling alignment of tens of thousands of sequences. Accuracy is lower than other methods but computation remains tractable.
Use PartTree only for datasets exceeding 10,000 sequences where other methods are too slow.
MAFFT auto-detects whether sequences are protein or nucleotide by examining character composition. Manual selection is useful for ambiguous cases like very short sequences.
For nucleotide alignments, MAFFT applies a different scoring matrix optimized for DNA/RNA. The distinction between DNA and RNA affects only the character set validation—alignment scoring is identical.
-. Compatible with FastTree and most downstream tools.The output alignment shows homologous positions as columns. Gap characters (-) indicate insertions or deletions relative to other sequences.
The Files tab includes the alignment and MAFFT's native diagnostic log. The log records the strategy that Auto actually selected, the runtime version, thread count, and any warnings produced during alignment.
Alignment length is the total number of columns, which exceeds any individual sequence length due to gaps. A good alignment minimizes scattered gaps and maximizes contiguous aligned blocks.
Conserved columns (identical residues across all sequences) suggest functional importance. Semi-conserved positions (similar amino acids) often indicate structural constraints.
Both tools produce high-quality alignments, but they excel in different scenarios:
MAFFT's iterative methods (L-INS-i, E-INS-i) generally achieve higher accuracy for difficult alignments with divergent sequences. The FFT-based approach is particularly effective when sequences have variable-length insertions.
Clustal Omega scales better to very large datasets (>10,000 sequences) thanks to its mBed algorithm. Its HMM-based profile alignment can be more robust for sequences with low complexity regions.
For most users with datasets under 1,000 sequences, either tool produces excellent results. We recommend trying both on a subset and comparing the alignments visually.
Multiple sequence alignment is typically the first step in analysis pipelines:
MAFFT assumes input sequences are homologous. It will produce an alignment for any input, but unrelated sequences yield meaningless results. Verify evolutionary relationships before aligning.
The iterative methods (L-INS-i, G-INS-i, E-INS-i) become slow for datasets exceeding a few hundred sequences. Switch to FFT-NS-2 or PartTree for larger datasets.
Very divergent sequences (below ~20% identity for proteins) may not align reliably with any sequence-based method. Consider structure-based alignment with USAlign for such cases.
Align protein, DNA, or RNA sequences with MUSCLE v5.3 using the native PPP or Super5 algorithm, with optional stratified and diversified ensembles.
Calculate batched Levenshtein distances and Needleman–Wunsch or Smith–Waterman sequence scores with StringZilla v5 CPU acceleration.
Ultra-fast sequence search and clustering. 10,000x faster than BLAST for database searches, with powerful sequence clustering capabilities for proteins and nucleotides.
USAlign (Universal Structure Alignment) aligns protein, RNA, and DNA structures to compute TM-scores and generate superposed structures. Compare 3D structures to assess structural similarity.
Perform multiple sequence alignment on protein or nucleotide sequences using the Clustal Omega algorithm.
Infer approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences.
Build phylogenetic trees using maximum likelihood with automatic model selection (ModelFinder) and ultrafast bootstrap support.
Rapidly align and compare DNA sequences using MUMmer4 nucmer. Perform pairwise genome comparisons to identify SNPs, indels, and structural variants between reference and query genomes.
Analyze immunoglobulin (antibody) and T cell receptor variable domain sequences. Identifies V/D/J gene segments, delineates CDR regions, and analyzes rearrangement junctions.
Fast protein structure search, comparison, and clustering. Search your structure against 200M+ AlphaFold predictions, compare 2 structures, or cluster up to 2500.