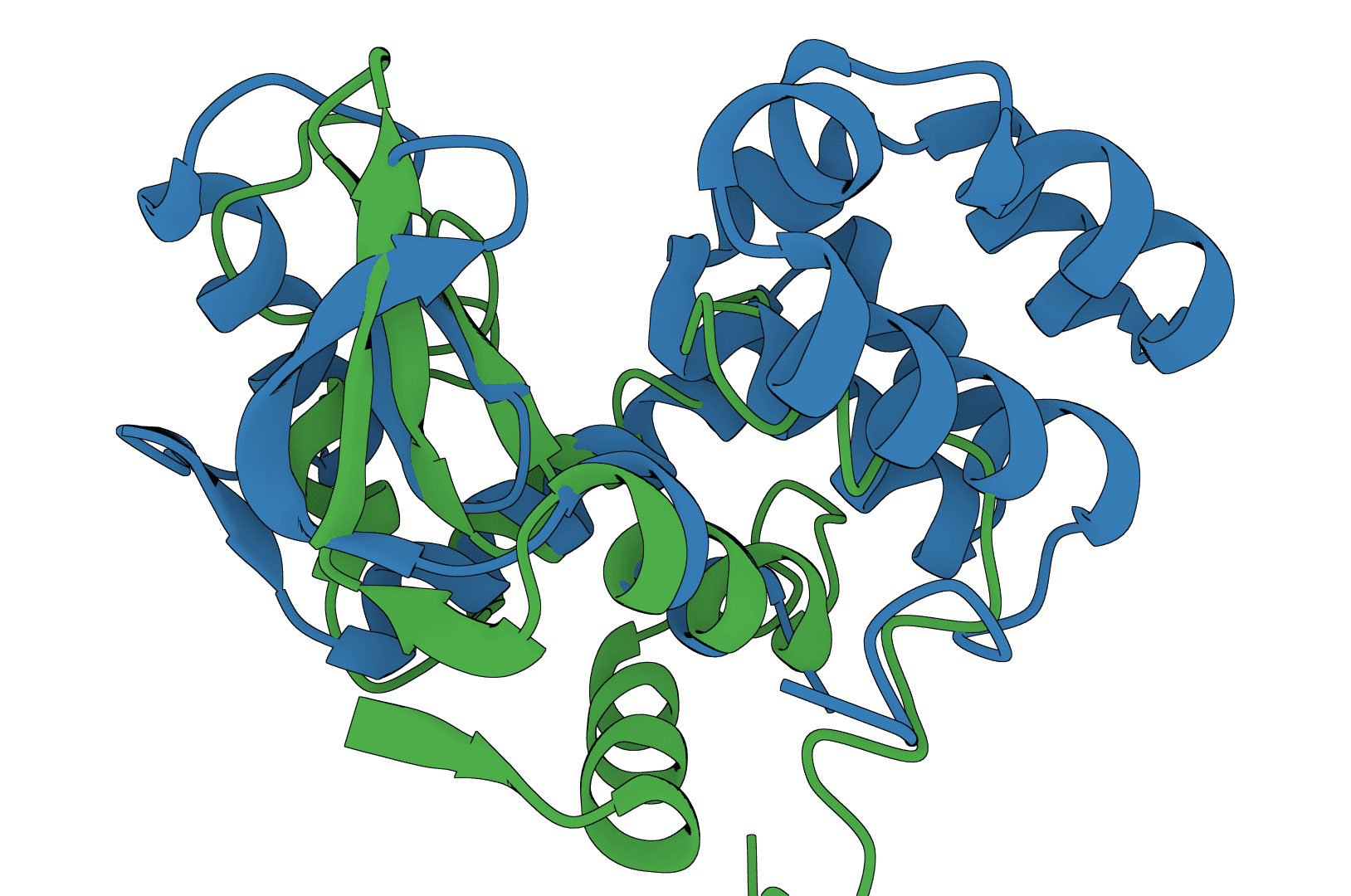
Universal structure alignment for proteins, RNA, and DNA molecules Learn more
Run
11 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job”.
Universal structure alignment for proteins, RNA, and DNA molecules Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
Universal structure alignment for proteins, RNA, and DNA molecules Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
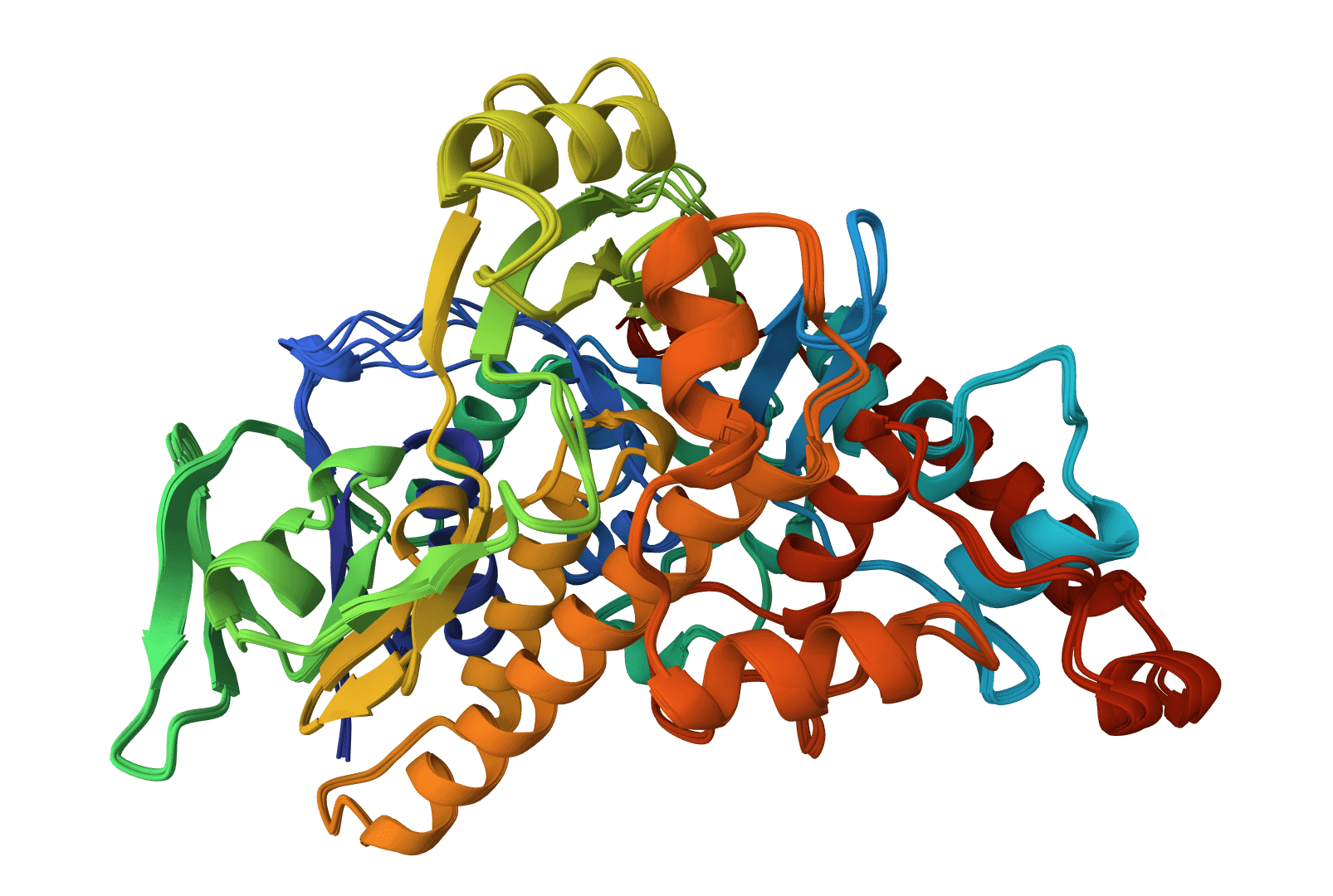
USAlign (Universal Structure Alignment) compares the 3D structures of proteins, RNA, and DNA molecules. Given two structures, it calculates a TM-score that quantifies their structural similarity and generates a superposed structure showing how they align in 3D space.
Structure alignment answers a fundamental question in structural biology: how similar are two molecular structures? This is essential for understanding evolutionary relationships, validating predicted structures against experimental ones, and identifying proteins with similar folds despite low sequence similarity. Unlike sequence alignment, structure alignment captures geometric similarity that persists even when sequences have diverged.
USAlign unifies several earlier algorithms (TM-align for proteins, RNA-align for RNA) into a single tool that handles any macromolecule type. For comparing predicted structures from tools like Boltz-2, ESMFold, or Chai-1 against experimental structures, USAlign provides the standard metrics used in the field.
USAlign finds the optimal superposition of two structures by maximizing the TM-score through iterative dynamic programming. The algorithm rotates and translates the mobile structure to best match the reference structure, then reports alignment quality metrics.
The TM-score measures global structural similarity on a scale from 0 to 1:
Where is the reference structure length, is the number of aligned residues, is the distance between aligned residue pairs after superposition, and is a length-dependent normalization factor.
The TM-score is length-independent, meaning it can fairly compare structures of different sizes. This distinguishes it from RMSD, which tends to increase with structure size even for similarly-shaped molecules.
| TM-score | Interpretation |
|---|---|
| Same fold (proteins) — structures share global topology | |
| Same fold (RNA) — nucleic acid structures share topology | |
| Similar fold with differences — related but distinct structures | |
| Different folds — unrelated structures |
For monomer alignments, USAlign reports two TM-scores: one normalized by structure 1's length and another by structure 2's length. For comparing a prediction to an experimental structure, use the score normalized by the experimental reference. Oligomer mode instead reports average TM-scores normalized by the shorter and longer complexes.
Root Mean Square Deviation (RMSD) measures the average distance between aligned atoms after optimal superposition:
RMSD is reported in Ångströms (Å). Lower values indicate better alignment. However, RMSD depends on the number of aligned residues—a partial alignment of a conserved core will have lower RMSD than a full-length alignment that includes flexible loops.
USAlign requires two structure files in PDB or mmCIF format. You can upload files directly or fetch structures from RCSB PDB using their 4-letter codes.
Structure 1 (Mobile): This structure will be rotated and translated to align with the reference. The output includes this structure after transformation.
Structure 2 (Reference): This structure remains fixed. TM-scores normalized by its length are typically preferred when comparing predictions to experimental structures.
Monomer: Aligns single chains. If your PDB contains multiple chains, only the first chain is used. This is the standard mode for comparing protein domains or single-chain structures.
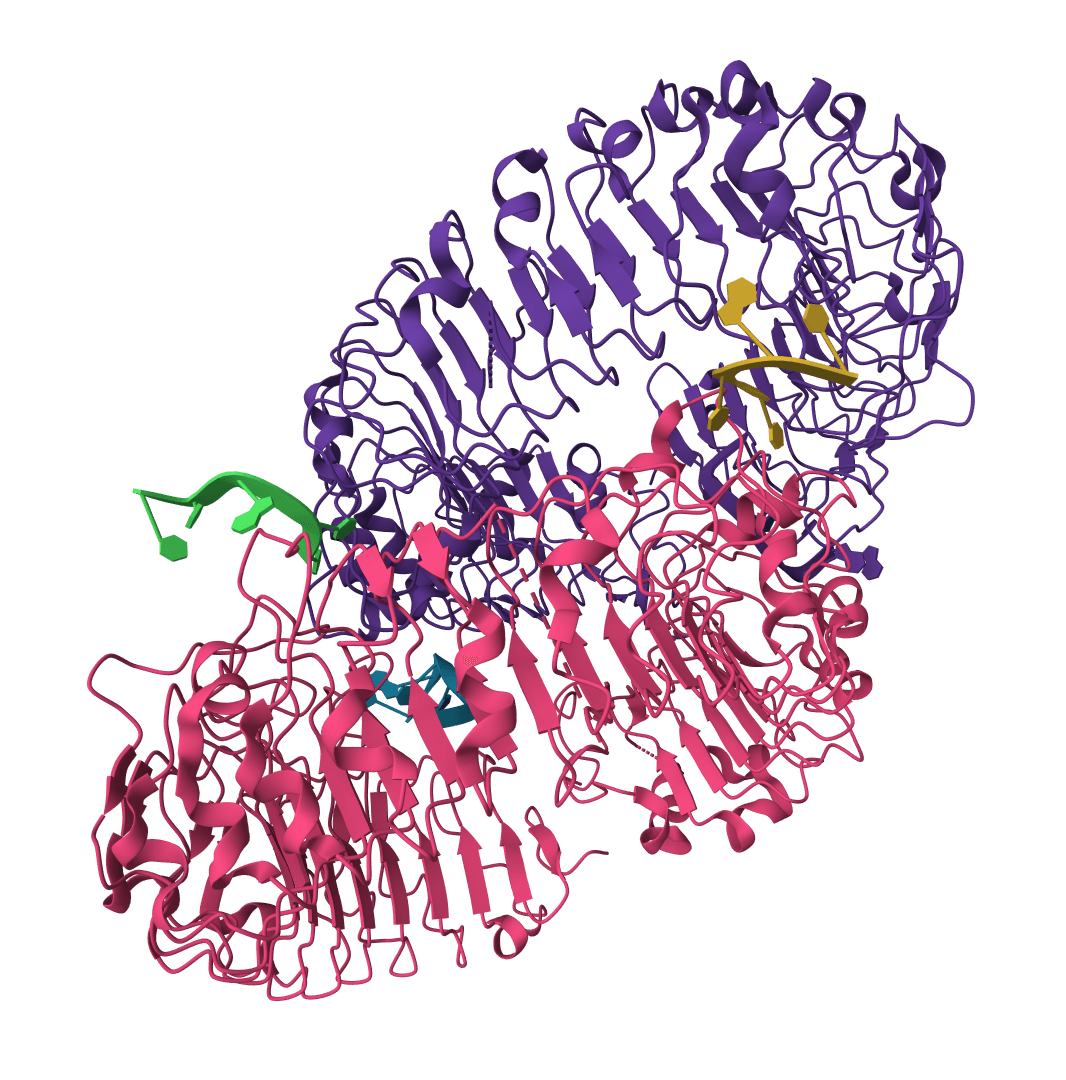
Oligomer: Aligns multi-chain complexes as a unit. Use this when comparing quaternary structures like dimers or larger assemblies where chain arrangement matters.
Circular permutation: Detects cases where the sequence has been circularly permuted—the N-terminus of one protein corresponds to an internal position in the other. This occurs in some protein families and can be missed by standard alignment.
Mirror image: Aligns mirror-image structures. Useful for detecting chirality-related relationships or artifacts in structure predictions.
Biological unit: Uses all chains in the structure file as they appear. Choose this for comparing complete biological assemblies.
All chains, first model: Uses every chain in the first model of each submitted coordinate file. This is the source setting commonly used for asymmetric-unit comparisons; it does not reduce the input to the first chain.
USAlign outputs several metrics alongside the superposed structure:
TM-score: The primary similarity metric. The same-fold reference threshold is 0.50 for proteins and 0.45 for RNA. Monomer results include both structure-length normalizations, while oligomer results include the native shorter- and longer-complex normalizations.
RMSD: Average atomic displacement after alignment, in Ångströms. Context matters: 2Å RMSD is excellent for a 200-residue protein but mediocre for a 50-residue domain.
Aligned length: Number of residue pairs included in the structural alignment. A partial alignment (e.g., 80 of 150 residues) suggests divergent regions that couldn't be matched.
Sequence identity: Percentage of aligned positions with identical amino acids. Low sequence identity (~20%) with high TM-score (~0.7) indicates remote homologs with conserved structure but diverged sequences.
The result files also include the complete native USAlign text report, all generated coordinate variants, source-generated PyMOL or ChimeraX scripts, and JSON run provenance. The visualization scripts use the downloadable result filenames so they can be run alongside the downloaded structures.
When validating structure predictions, always use the experimental structure as the reference (Structure 2). This ensures TM-score normalization reflects how well the prediction captures the known structure.
For multi-domain proteins, consider aligning domains separately. A full-length alignment may show poor scores if domain orientations differ, even when individual domains are well-predicted.
Compare RMSD in context. Global RMSD includes all aligned residues, but flexible loops and termini often contribute disproportionately. Many publications report core RMSD (excluding flexible regions) alongside global RMSD.
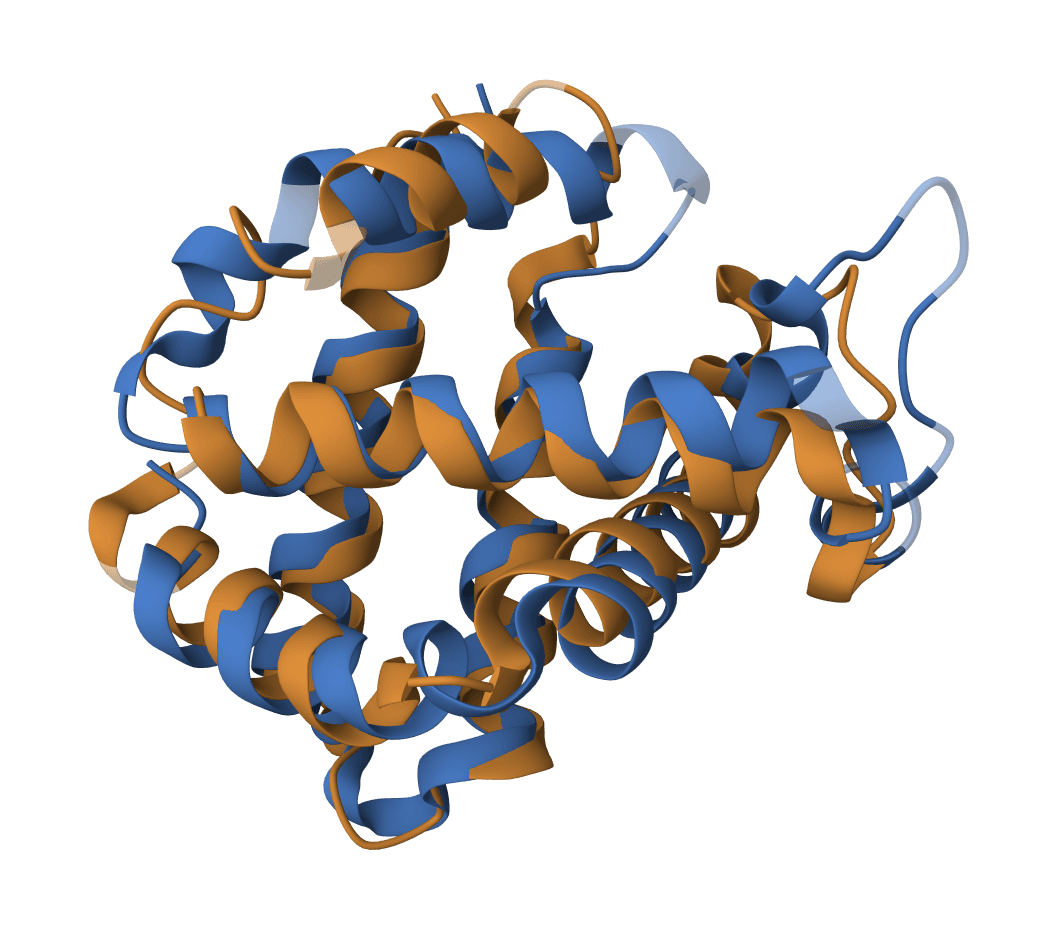
Perform multiple sequence alignment using MAFFT (Multiple Alignment using Fast Fourier Transform). Supports multiple algorithms from fast progressive to highly accurate iterative methods.
Align protein, DNA, or RNA sequences with MUSCLE v5.3 using the native PPP or Super5 algorithm, with optional stratified and diversified ensembles.
Calculate batched Levenshtein distances and Needleman–Wunsch or Smith–Waterman sequence scores with StringZilla v5 CPU acceleration.
Perform multiple sequence alignment on protein or nucleotide sequences using the Clustal Omega algorithm.
Infer approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences.
Fast protein structure search, comparison, and clustering. Search your structure against 200M+ AlphaFold predictions, compare 2 structures, or cluster up to 2500.
Build phylogenetic trees using maximum likelihood with automatic model selection (ModelFinder) and ultrafast bootstrap support.
Ultra-fast sequence search and clustering. 10,000x faster than BLAST for database searches, with powerful sequence clustering capabilities for proteins and nucleotides.
Rapidly align and compare DNA sequences using MUMmer4 nucmer. Perform pairwise genome comparisons to identify SNPs, indels, and structural variants between reference and query genomes.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
USAlign (Universal Structure Alignment) compares the 3D structures of proteins, RNA, and DNA molecules. Given two structures, it calculates a TM-score that quantifies their structural similarity and generates a superposed structure showing how they align in 3D space.
Structure alignment answers a fundamental question in structural biology: how similar are two molecular structures? This is essential for understanding evolutionary relationships, validating predicted structures against experimental ones, and identifying proteins with similar folds despite low sequence similarity. Unlike sequence alignment, structure alignment captures geometric similarity that persists even when sequences have diverged.
USAlign unifies several earlier algorithms (TM-align for proteins, RNA-align for RNA) into a single tool that handles any macromolecule type. For comparing predicted structures from tools like Boltz-2, ESMFold, or Chai-1 against experimental structures, USAlign provides the standard metrics used in the field.
USAlign finds the optimal superposition of two structures by maximizing the TM-score through iterative dynamic programming. The algorithm rotates and translates the mobile structure to best match the reference structure, then reports alignment quality metrics.
The TM-score measures global structural similarity on a scale from 0 to 1:
Where is the reference structure length, is the number of aligned residues, is the distance between aligned residue pairs after superposition, and is a length-dependent normalization factor.
The TM-score is length-independent, meaning it can fairly compare structures of different sizes. This distinguishes it from RMSD, which tends to increase with structure size even for similarly-shaped molecules.
| TM-score | Interpretation |
|---|---|
| Same fold (proteins) — structures share global topology | |
| Same fold (RNA) — nucleic acid structures share topology | |
| Similar fold with differences — related but distinct structures | |
| Different folds — unrelated structures |
For monomer alignments, USAlign reports two TM-scores: one normalized by structure 1's length and another by structure 2's length. For comparing a prediction to an experimental structure, use the score normalized by the experimental reference. Oligomer mode instead reports average TM-scores normalized by the shorter and longer complexes.
Root Mean Square Deviation (RMSD) measures the average distance between aligned atoms after optimal superposition:
RMSD is reported in Ångströms (Å). Lower values indicate better alignment. However, RMSD depends on the number of aligned residues—a partial alignment of a conserved core will have lower RMSD than a full-length alignment that includes flexible loops.
USAlign requires two structure files in PDB or mmCIF format. You can upload files directly or fetch structures from RCSB PDB using their 4-letter codes.
Structure 1 (Mobile): This structure will be rotated and translated to align with the reference. The output includes this structure after transformation.
Structure 2 (Reference): This structure remains fixed. TM-scores normalized by its length are typically preferred when comparing predictions to experimental structures.
Monomer: Aligns single chains. If your PDB contains multiple chains, only the first chain is used. This is the standard mode for comparing protein domains or single-chain structures.
Oligomer: Aligns multi-chain complexes as a unit. Use this when comparing quaternary structures like dimers or larger assemblies where chain arrangement matters.
Circular permutation: Detects cases where the sequence has been circularly permuted—the N-terminus of one protein corresponds to an internal position in the other. This occurs in some protein families and can be missed by standard alignment.
Mirror image: Aligns mirror-image structures. Useful for detecting chirality-related relationships or artifacts in structure predictions.
Biological unit: Uses all chains in the structure file as they appear. Choose this for comparing complete biological assemblies.
All chains, first model: Uses every chain in the first model of each submitted coordinate file. This is the source setting commonly used for asymmetric-unit comparisons; it does not reduce the input to the first chain.
USAlign outputs several metrics alongside the superposed structure:
TM-score: The primary similarity metric. The same-fold reference threshold is 0.50 for proteins and 0.45 for RNA. Monomer results include both structure-length normalizations, while oligomer results include the native shorter- and longer-complex normalizations.
RMSD: Average atomic displacement after alignment, in Ångströms. Context matters: 2Å RMSD is excellent for a 200-residue protein but mediocre for a 50-residue domain.
Aligned length: Number of residue pairs included in the structural alignment. A partial alignment (e.g., 80 of 150 residues) suggests divergent regions that couldn't be matched.
Sequence identity: Percentage of aligned positions with identical amino acids. Low sequence identity (~20%) with high TM-score (~0.7) indicates remote homologs with conserved structure but diverged sequences.
The result files also include the complete native USAlign text report, all generated coordinate variants, source-generated PyMOL or ChimeraX scripts, and JSON run provenance. The visualization scripts use the downloadable result filenames so they can be run alongside the downloaded structures.
When validating structure predictions, always use the experimental structure as the reference (Structure 2). This ensures TM-score normalization reflects how well the prediction captures the known structure.
For multi-domain proteins, consider aligning domains separately. A full-length alignment may show poor scores if domain orientations differ, even when individual domains are well-predicted.
Compare RMSD in context. Global RMSD includes all aligned residues, but flexible loops and termini often contribute disproportionately. Many publications report core RMSD (excluding flexible regions) alongside global RMSD.
Perform multiple sequence alignment using MAFFT (Multiple Alignment using Fast Fourier Transform). Supports multiple algorithms from fast progressive to highly accurate iterative methods.
Align protein, DNA, or RNA sequences with MUSCLE v5.3 using the native PPP or Super5 algorithm, with optional stratified and diversified ensembles.
Calculate batched Levenshtein distances and Needleman–Wunsch or Smith–Waterman sequence scores with StringZilla v5 CPU acceleration.
Perform multiple sequence alignment on protein or nucleotide sequences using the Clustal Omega algorithm.
Infer approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences.
Fast protein structure search, comparison, and clustering. Search your structure against 200M+ AlphaFold predictions, compare 2 structures, or cluster up to 2500.
Build phylogenetic trees using maximum likelihood with automatic model selection (ModelFinder) and ultrafast bootstrap support.
Ultra-fast sequence search and clustering. 10,000x faster than BLAST for database searches, with powerful sequence clustering capabilities for proteins and nucleotides.
Rapidly align and compare DNA sequences using MUMmer4 nucmer. Perform pairwise genome comparisons to identify SNPs, indels, and structural variants between reference and query genomes.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.