Run
50 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
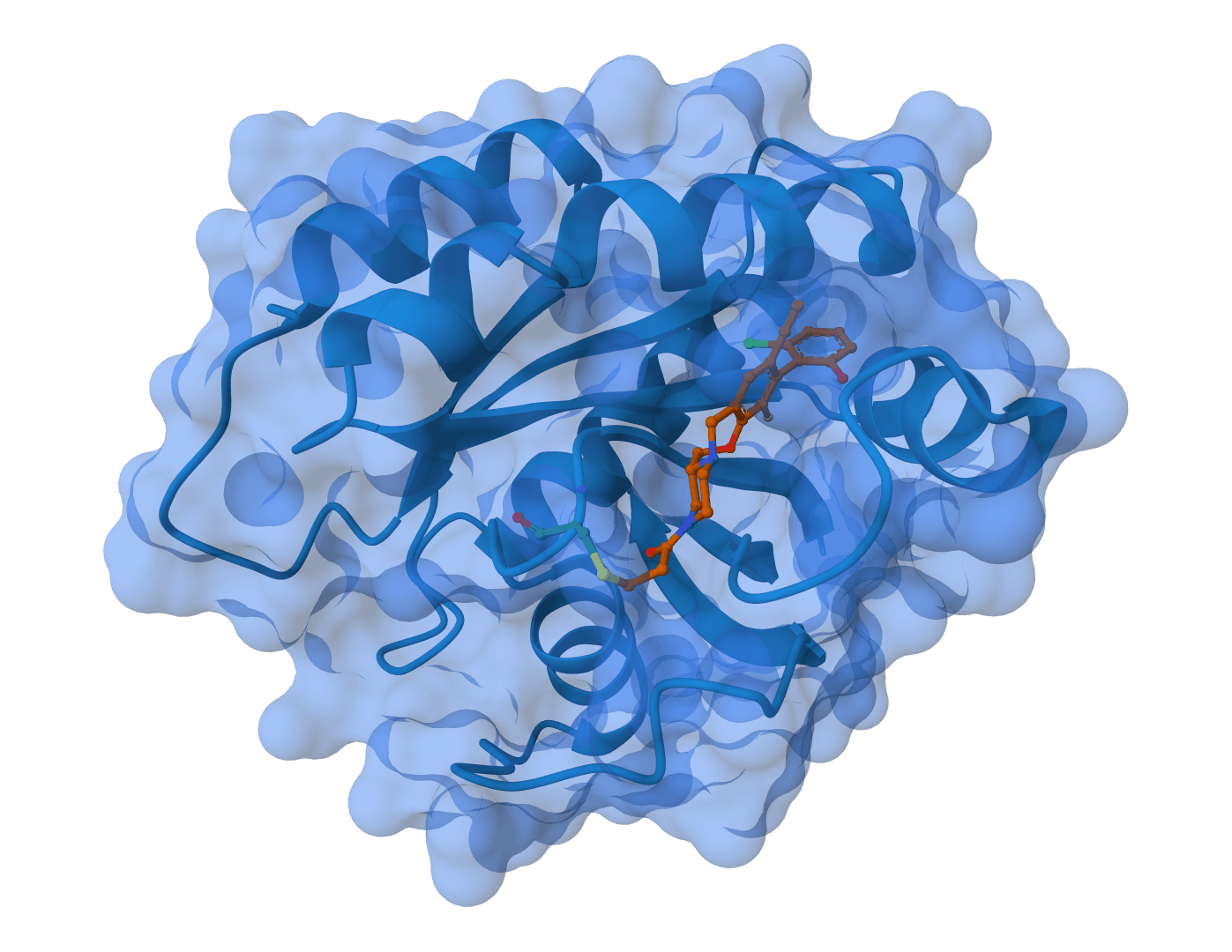
Human KRAS G12C protein with covalent ligand
A transcription factor and DNA complex
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Human KRAS G12C protein with covalent ligand
A transcription factor and DNA complex
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Human KRAS G12C protein with covalent ligand
A transcription factor and DNA complex
Boltz-2 is a biomolecular foundation model that jointly predicts molecular complex structures and protein-ligand binding affinities. Developed by MIT Jameel Clinic and Recursion and released in June 2025, it extends Boltz-1—the most widely used open-source alternative to AlphaFold3 across academia and industry.
The model achieves binding affinity predictions approaching the accuracy of physics-based free-energy perturbation (FEP) calculations while running over 1000x faster. Traditional FEP methods cost hundreds of dollars per prediction and require 6–12 hours to complete; Boltz-2 achieves comparable accuracy in approximately 20 seconds on a single GPU.
Boltz-2 supports multi-component biomolecular complexes including proteins, small molecule ligands, DNA, and RNA. This broad coverage makes it particularly valuable for drug discovery workflows requiring rapid screening of compound libraries before experimental synthesis.
ProteinIQ hosts Boltz-2 on GPU infrastructure, delivering structure and affinity predictions through a browser interface without local installation or command-line configuration.
Boltz-2 accepts multiple molecule types that combine into a single prediction job. Chain IDs (A, B, C...) are assigned automatically and displayed in the interface—these identifiers are needed when defining constraints.
| Input | Description |
|---|---|
Protein | FASTA sequences, PDB structure files, or RCSB chain fetches. Optional A3M pre-computed MSA files can be uploaded directly on each protein input. CIF/mmCIF protein inputs are not supported; use CIF files as templates instead. Up to 10 chains. |
Ligand (SMILES) | SMILES strings, SDF/MOL/MOL2 files, or PubChem compound IDs. Up to 10 ligands. |
Ligand (CCD) | Standard codes from the PDB Chemical Component Dictionary (e.g., ATP, NAD, HEM). |
DNA | FASTA sequences supplied as text or file input. Up to 10 chains. |
RNA | FASTA sequences supplied as text or file input. Up to 10 chains. |
Template | Known structures used to bias prediction toward a homologous fold. Templates are separate structural guide inputs, not extra sequence chains. PDB/CIF files or RCSB structure IDs. Up to 5 templates. |
| Setting | Description |
|---|---|
Number of samples | Structure predictions to generate (1–20, default 1). More samples capture conformational diversity but increase runtime. |
Confidence threshold | Minimum confidence score for predictions (0.0–1.0, default 0.5). Higher values filter low-confidence results. |
Multiple sequence alignment (MSA) aligns a target protein sequence against thousands of evolutionarily related sequences. By analyzing coevolution patterns—which amino acid positions change together across evolution—the model infers spatial relationships in the 3D structure.
For custom alignments, upload an A3M file directly on the matching protein input. Copied identical proteins share that protein input's MSA. When automatic MSA generation is enabled, provide custom MSAs for all protein inputs or leave all protein inputs without custom MSAs.
| Setting | Description |
|---|---|
Generate MSA | Off by default. Enable automatic MSA generation via ColabFold when evolutionary context may improve a protein prediction. Leave it off for faster single-sequence inference or synthetic proteins without natural homologs. |
MSA depth | Search thoroughness: Shallow (2048 seqs, fast), Normal (8192 seqs, default), or Deep (16384 seqs, slow but thorough). |
MSA pairing strategy | How MSAs from different chains combine: Greedy (default) or Complete (slower, better for obligate heterodimers). |
Max MSA sequences | Maximum aligned sequences (512–16384, default 8192). Lower values are faster; higher values may improve accuracy. |
Subsample MSA | Reduces a large alignment to a smaller working set for faster inference on long proteins. |
Subsampled sequences | Number of sequences retained after subsampling (256–4096, default 1024). Used only when Subsample MSA is enabled. |
MSA server URL | Optional custom endpoint for external ColabFold-compatible MSA services. |
MSA username | Optional basic-auth username for a private MSA server. Must be paired with MSA password. |
MSA password | Optional basic-auth password for a private MSA server. |
MSA API key header | Custom header name for token-based authentication to an MSA service. |
MSA API key value | Token value sent with the configured API key header. |
Boltz-2 uses diffusion-based structure generation with iterative refinement ("recycling"). These parameters control that process.
| Setting | Description |
|---|---|
Recycling steps | Refinement iterations (1–10, default 3). More steps improve accuracy at increased runtime. |
Sampling steps | Diffusion denoising steps (50–500, default 200). Higher values produce more refined structures. |
Step scale | Temperature controlling sampling diversity (1.0–2.0, default 1.5). Higher values explore more conformational space. |
Output format | CIF (recommended) or PDB (legacy compatibility). |
MW-corrected affinity | Applies molecular weight correction when comparing ligands of different sizes. |
Affinity sampling steps | Diffusion steps for affinity prediction (50–500, default 200). |
Affinity samples | Independent affinity predictions to average (1–20, default 5). More samples reduce variance. |
Save PAE matrix | Exports the full predicted aligned error matrix for interface and domain-quality analysis. |
Save PDE matrix | Exports the predicted distance error matrix for downstream structural inspection. |
Random seed | Fixes stochastic sampling for reproducible reruns. Leaving the field empty uses a fresh random seed. |
Model version | Chooses Boltz-2 for joint structure and affinity prediction or Boltz-1 for legacy structure-only inference. |
Constraints guide predictions toward specific structural features when prior knowledge exists about the system, including crystallography, mutagenesis studies, or crosslinking mass spectrometry data.
| Setting | Description |
|---|---|
Use constraint potentials | Adds inference-time potentials for pocket and contact constraints marked force=true. Constraints are still passed to Boltz when this setting is off. |
Pocket constraints | Guides a ligand toward specified residues. Format: binder|contacts|max_distance|force (for example, C|A:45,A:46|6.0|true). |
Covalent bonds | Defines bonds between canonical polymer residues and Ligand (CCD) inputs. Custom SMILES, MOL, SDF, and MOL2 ligands are not supported. Use standardized CCD atom names from the component CIF on RCSB. Format: chain:residue:atom,chain:residue:atom (for example, A:12:SG,B:1:C22). |
Contact constraints | Guides two residues or atoms within a specified distance. Format: chain:residue,chain:residue|max_distance|force. |
Templates can be used either as soft structural priors or as stronger geometric constraints for cases where a homologous backbone is already known.
| Setting | Description |
|---|---|
Enforce template backbone | Keeps the predicted backbone close to the supplied template instead of treating the template as loose guidance. |
Template deviation threshold (Å) | Maximum allowed backbone deviation from the template when enforcement is enabled. |
Template chain mapping | Assigns template indices to specific query chains. Format: template_index:chain_ids. |
| Setting | Description |
|---|---|
Copies | Number of identical copies for the selected molecule. Copies are assigned consecutive chain IDs and submitted using Boltz's native identical-entity format. |
Cyclic | Marks the selected protein, DNA, or RNA molecule as head-to-tail cyclic. |
Add modification | Adds a residue modification for the selected protein, DNA, or RNA molecule using a residue position and CCD code. Common codes include SEP (phosphoserine), TPO (phosphothreonine), and MLY (methylated lysine). |
Boltz-2 returns ranked predictions, each with downloadable structure files and a data table summarizing structure and affinity outputs. Optional diagnostic matrices can also be saved from the advanced settings.
| Column | Description |
|---|---|
Rank | Prediction ranking by confidence. |
Affinity (log10 IC50) | Quantitative binding strength in log10(IC50) μM. More negative = stronger binding. |
Confidence | Structural prediction confidence (0–1). |
Structure file | Predicted complex saved as CIF or PDB, depending on the selected output format. |
PAE/PDE files | Optional error matrices written when Save PAE matrix or Save PDE matrix is enabled. |
Boltz-2 outputs two affinity metrics serving different purposes:
Affinity probability (0–1): Binary classifier confidence that the molecule binds. Values above 0.7 indicate strong predicted binders; below 0.5 suggests unlikely binding. Best used for initial screening to separate hits from non-binders.
Affinity value (log10 IC50 in μM): Quantitative binding strength. More negative values indicate stronger binding.
| Value | Interpretation | Typical use case |
|---|---|---|
| < −8 | Very strong (nM) | Clinical candidates |
| −8 to −6 | Strong (low μM) | Lead compounds |
| −6 to −4 | Moderate | Hit compounds |
| > −4 | Weak | Likely inactive |
Confidence reflects certainty about the predicted structure, not the affinity estimate. Low confidence does not mean the prediction is wrong—it indicates the result warrants closer examination.
| Score | Interpretation |
|---|---|
| > 0.7 | High confidence—prediction likely reliable |
| 0.5–0.7 | Moderate—visual inspection recommended |
| < 0.5 | Low—may indicate a difficult target, missing MSA data, or unusual binding mode |
Boltz-2 extends Boltz-1 with an affinity prediction module trained on millions of binding measurements. The result is a single model predicting both 3D structures and binding strength.
The model processes input in two stages. First, the structure module generates 3D coordinates using diffusion—starting from noise and iteratively refining toward a physically plausible structure. This approach parallels image generation models but operates on molecular geometry.
Second, the affinity module takes the predicted structure and outputs binding predictions: both a binary "does it bind?" probability and a quantitative IC50 estimate.
Boltz-2 was trained on approximately 5 million binding affinity measurements (Kd and IC50 values) from public assays, plus molecular dynamics simulations capturing protein flexibility.
The training data spans diverse complex types: protein-ligand, protein-DNA, protein-RNA, and protein-protein interactions. This breadth accounts for Boltz-2's effectiveness on multi-molecule complexes.
For structure prediction, Boltz-2 matches or slightly outperforms AlphaFold3, Chai-1, and Boltz-1 on recent PDB deposits (2024–2025), with particular strength on RNA, DNA-protein complexes, and antibody-antigen interactions.
For affinity prediction, Boltz-2 achieves Pearson correlation r ≈ 0.62 on the FEP+ benchmark—matching physics-based free energy perturbation methods that require hours instead of seconds. In the CASP16 affinity challenge, it outperformed all submitted methods across 140 complexes.
Boltz-2 excels in scenarios requiring both structure and affinity prediction in a single workflow.
| Scenario | Recommendation |
|---|---|
| Need structure + quantitative affinity | Boltz-2 |
| Multi-component complexes (protein + DNA + ligand) | Boltz-2 |
| Already have protein structure, need ligand poses only | DiffDock (faster, no affinity) |
| High-throughput screening with known binding site | AutoDock Vina or AutoDock-GPU |
| CNN-enhanced scoring on traditional docking | GNINA |
| Feature | Boltz-2 | DiffDock | AutoDock Vina | FEP+ |
|---|---|---|---|---|
| Predicts structure | Yes | No (pose only) | No (pose only) | No |
| Predicts affinity | Yes (quantitative) | No | Yes (scoring) | Yes (quantitative) |
| Affinity accuracy | r ≈ 0.62 | N/A | r ≈ 0.3–0.4 | r ≈ 0.6–0.7 |
| Speed | ~20 sec | ~30 sec | ~1 min | 6–12 hours |
| Molecule types | Protein, DNA, RNA, ligand | Protein, ligand | Protein, ligand | Protein, ligand |
r ≈ 0.35–0.37). Synthetic or designed proteins lacking natural homologs may yield lower-confidence results.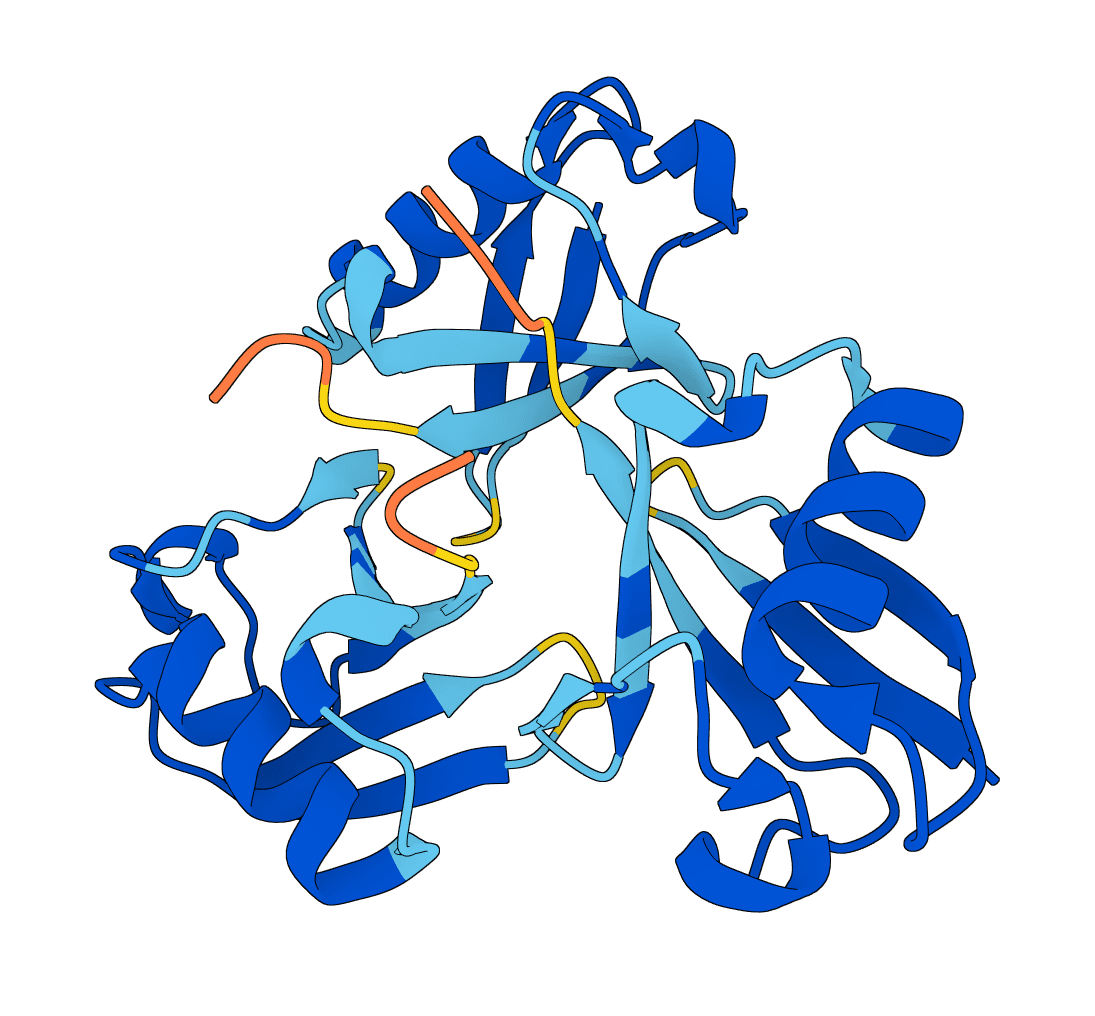
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
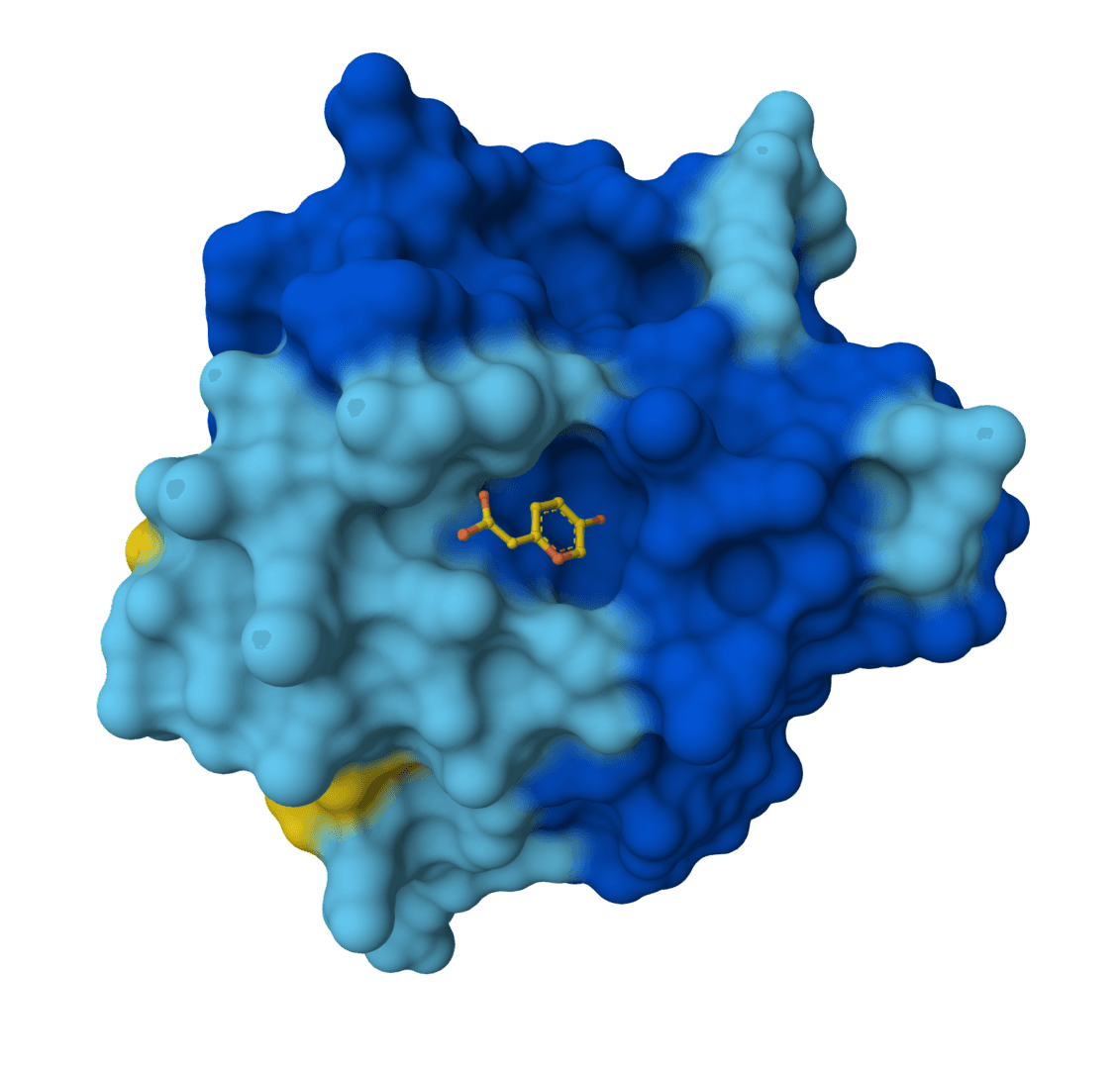
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
OpenFold-3 is an open-source AI model for biomolecular structure prediction, aiming to reproduce AlphaFold3. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Enhanced Protenix v2 biomolecular structure prediction by ByteDance. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Open-source structure prediction neural network for proteins, nucleic acids, and small molecules. State-of-the-art accuracy with multi-chain support.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
Boltz-2 is a biomolecular foundation model that jointly predicts molecular complex structures and protein-ligand binding affinities. Developed by MIT Jameel Clinic and Recursion and released in June 2025, it extends Boltz-1—the most widely used open-source alternative to AlphaFold3 across academia and industry.
The model achieves binding affinity predictions approaching the accuracy of physics-based free-energy perturbation (FEP) calculations while running over 1000x faster. Traditional FEP methods cost hundreds of dollars per prediction and require 6–12 hours to complete; Boltz-2 achieves comparable accuracy in approximately 20 seconds on a single GPU.
Boltz-2 supports multi-component biomolecular complexes including proteins, small molecule ligands, DNA, and RNA. This broad coverage makes it particularly valuable for drug discovery workflows requiring rapid screening of compound libraries before experimental synthesis.
ProteinIQ hosts Boltz-2 on GPU infrastructure, delivering structure and affinity predictions through a browser interface without local installation or command-line configuration.
Boltz-2 accepts multiple molecule types that combine into a single prediction job. Chain IDs (A, B, C...) are assigned automatically and displayed in the interface—these identifiers are needed when defining constraints.
| Input | Description |
|---|---|
Protein | FASTA sequences, PDB structure files, or RCSB chain fetches. Optional A3M pre-computed MSA files can be uploaded directly on each protein input. CIF/mmCIF protein inputs are not supported; use CIF files as templates instead. Up to 10 chains. |
Ligand (SMILES) | SMILES strings, SDF/MOL/MOL2 files, or PubChem compound IDs. Up to 10 ligands. |
Ligand (CCD) | Standard codes from the PDB Chemical Component Dictionary (e.g., ATP, NAD, HEM). |
DNA | FASTA sequences supplied as text or file input. Up to 10 chains. |
RNA | FASTA sequences supplied as text or file input. Up to 10 chains. |
Template | Known structures used to bias prediction toward a homologous fold. Templates are separate structural guide inputs, not extra sequence chains. PDB/CIF files or RCSB structure IDs. Up to 5 templates. |
| Setting | Description |
|---|---|
Number of samples | Structure predictions to generate (1–20, default 1). More samples capture conformational diversity but increase runtime. |
Confidence threshold | Minimum confidence score for predictions (0.0–1.0, default 0.5). Higher values filter low-confidence results. |
Multiple sequence alignment (MSA) aligns a target protein sequence against thousands of evolutionarily related sequences. By analyzing coevolution patterns—which amino acid positions change together across evolution—the model infers spatial relationships in the 3D structure.
For custom alignments, upload an A3M file directly on the matching protein input. Copied identical proteins share that protein input's MSA. When automatic MSA generation is enabled, provide custom MSAs for all protein inputs or leave all protein inputs without custom MSAs.
| Setting | Description |
|---|---|
Generate MSA | Off by default. Enable automatic MSA generation via ColabFold when evolutionary context may improve a protein prediction. Leave it off for faster single-sequence inference or synthetic proteins without natural homologs. |
MSA depth | Search thoroughness: Shallow (2048 seqs, fast), Normal (8192 seqs, default), or Deep (16384 seqs, slow but thorough). |
MSA pairing strategy | How MSAs from different chains combine: Greedy (default) or Complete (slower, better for obligate heterodimers). |
Max MSA sequences | Maximum aligned sequences (512–16384, default 8192). Lower values are faster; higher values may improve accuracy. |
Subsample MSA | Reduces a large alignment to a smaller working set for faster inference on long proteins. |
Subsampled sequences | Number of sequences retained after subsampling (256–4096, default 1024). Used only when Subsample MSA is enabled. |
MSA server URL | Optional custom endpoint for external ColabFold-compatible MSA services. |
MSA username | Optional basic-auth username for a private MSA server. Must be paired with MSA password. |
MSA password | Optional basic-auth password for a private MSA server. |
MSA API key header | Custom header name for token-based authentication to an MSA service. |
MSA API key value | Token value sent with the configured API key header. |
Boltz-2 uses diffusion-based structure generation with iterative refinement ("recycling"). These parameters control that process.
| Setting | Description |
|---|---|
Recycling steps | Refinement iterations (1–10, default 3). More steps improve accuracy at increased runtime. |
Sampling steps | Diffusion denoising steps (50–500, default 200). Higher values produce more refined structures. |
Step scale | Temperature controlling sampling diversity (1.0–2.0, default 1.5). Higher values explore more conformational space. |
Output format | CIF (recommended) or PDB (legacy compatibility). |
MW-corrected affinity | Applies molecular weight correction when comparing ligands of different sizes. |
Affinity sampling steps | Diffusion steps for affinity prediction (50–500, default 200). |
Affinity samples | Independent affinity predictions to average (1–20, default 5). More samples reduce variance. |
Save PAE matrix | Exports the full predicted aligned error matrix for interface and domain-quality analysis. |
Save PDE matrix | Exports the predicted distance error matrix for downstream structural inspection. |
Random seed | Fixes stochastic sampling for reproducible reruns. Leaving the field empty uses a fresh random seed. |
Model version | Chooses Boltz-2 for joint structure and affinity prediction or Boltz-1 for legacy structure-only inference. |
Constraints guide predictions toward specific structural features when prior knowledge exists about the system, including crystallography, mutagenesis studies, or crosslinking mass spectrometry data.
| Setting | Description |
|---|---|
Use constraint potentials | Adds inference-time potentials for pocket and contact constraints marked force=true. Constraints are still passed to Boltz when this setting is off. |
Pocket constraints | Guides a ligand toward specified residues. Format: binder|contacts|max_distance|force (for example, C|A:45,A:46|6.0|true). |
Covalent bonds | Defines bonds between canonical polymer residues and Ligand (CCD) inputs. Custom SMILES, MOL, SDF, and MOL2 ligands are not supported. Use standardized CCD atom names from the component CIF on RCSB. Format: chain:residue:atom,chain:residue:atom (for example, A:12:SG,B:1:C22). |
Contact constraints | Guides two residues or atoms within a specified distance. Format: chain:residue,chain:residue|max_distance|force. |
Templates can be used either as soft structural priors or as stronger geometric constraints for cases where a homologous backbone is already known.
| Setting | Description |
|---|---|
Enforce template backbone | Keeps the predicted backbone close to the supplied template instead of treating the template as loose guidance. |
Template deviation threshold (Å) | Maximum allowed backbone deviation from the template when enforcement is enabled. |
Template chain mapping | Assigns template indices to specific query chains. Format: template_index:chain_ids. |
| Setting | Description |
|---|---|
Copies | Number of identical copies for the selected molecule. Copies are assigned consecutive chain IDs and submitted using Boltz's native identical-entity format. |
Cyclic | Marks the selected protein, DNA, or RNA molecule as head-to-tail cyclic. |
Add modification | Adds a residue modification for the selected protein, DNA, or RNA molecule using a residue position and CCD code. Common codes include SEP (phosphoserine), TPO (phosphothreonine), and MLY (methylated lysine). |
Boltz-2 returns ranked predictions, each with downloadable structure files and a data table summarizing structure and affinity outputs. Optional diagnostic matrices can also be saved from the advanced settings.
| Column | Description |
|---|---|
Rank | Prediction ranking by confidence. |
Affinity (log10 IC50) | Quantitative binding strength in log10(IC50) μM. More negative = stronger binding. |
Confidence | Structural prediction confidence (0–1). |
Structure file | Predicted complex saved as CIF or PDB, depending on the selected output format. |
PAE/PDE files | Optional error matrices written when Save PAE matrix or Save PDE matrix is enabled. |
Boltz-2 outputs two affinity metrics serving different purposes:
Affinity probability (0–1): Binary classifier confidence that the molecule binds. Values above 0.7 indicate strong predicted binders; below 0.5 suggests unlikely binding. Best used for initial screening to separate hits from non-binders.
Affinity value (log10 IC50 in μM): Quantitative binding strength. More negative values indicate stronger binding.
| Value | Interpretation | Typical use case |
|---|---|---|
| < −8 | Very strong (nM) | Clinical candidates |
| −8 to −6 | Strong (low μM) | Lead compounds |
| −6 to −4 | Moderate | Hit compounds |
| > −4 | Weak | Likely inactive |
Confidence reflects certainty about the predicted structure, not the affinity estimate. Low confidence does not mean the prediction is wrong—it indicates the result warrants closer examination.
| Score | Interpretation |
|---|---|
| > 0.7 | High confidence—prediction likely reliable |
| 0.5–0.7 | Moderate—visual inspection recommended |
| < 0.5 | Low—may indicate a difficult target, missing MSA data, or unusual binding mode |
Boltz-2 extends Boltz-1 with an affinity prediction module trained on millions of binding measurements. The result is a single model predicting both 3D structures and binding strength.
The model processes input in two stages. First, the structure module generates 3D coordinates using diffusion—starting from noise and iteratively refining toward a physically plausible structure. This approach parallels image generation models but operates on molecular geometry.
Second, the affinity module takes the predicted structure and outputs binding predictions: both a binary "does it bind?" probability and a quantitative IC50 estimate.
Boltz-2 was trained on approximately 5 million binding affinity measurements (Kd and IC50 values) from public assays, plus molecular dynamics simulations capturing protein flexibility.
The training data spans diverse complex types: protein-ligand, protein-DNA, protein-RNA, and protein-protein interactions. This breadth accounts for Boltz-2's effectiveness on multi-molecule complexes.
For structure prediction, Boltz-2 matches or slightly outperforms AlphaFold3, Chai-1, and Boltz-1 on recent PDB deposits (2024–2025), with particular strength on RNA, DNA-protein complexes, and antibody-antigen interactions.
For affinity prediction, Boltz-2 achieves Pearson correlation r ≈ 0.62 on the FEP+ benchmark—matching physics-based free energy perturbation methods that require hours instead of seconds. In the CASP16 affinity challenge, it outperformed all submitted methods across 140 complexes.
Boltz-2 excels in scenarios requiring both structure and affinity prediction in a single workflow.
| Scenario | Recommendation |
|---|---|
| Need structure + quantitative affinity | Boltz-2 |
| Multi-component complexes (protein + DNA + ligand) | Boltz-2 |
| Already have protein structure, need ligand poses only | DiffDock (faster, no affinity) |
| High-throughput screening with known binding site | AutoDock Vina or AutoDock-GPU |
| CNN-enhanced scoring on traditional docking | GNINA |
| Feature | Boltz-2 | DiffDock | AutoDock Vina | FEP+ |
|---|---|---|---|---|
| Predicts structure | Yes | No (pose only) | No (pose only) | No |
| Predicts affinity | Yes (quantitative) | No | Yes (scoring) | Yes (quantitative) |
| Affinity accuracy | r ≈ 0.62 | N/A | r ≈ 0.3–0.4 | r ≈ 0.6–0.7 |
| Speed | ~20 sec | ~30 sec | ~1 min | 6–12 hours |
| Molecule types | Protein, DNA, RNA, ligand | Protein, ligand | Protein, ligand | Protein, ligand |
r ≈ 0.35–0.37). Synthetic or designed proteins lacking natural homologs may yield lower-confidence results.LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
OpenFold-3 is an open-source AI model for biomolecular structure prediction, aiming to reproduce AlphaFold3. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Enhanced Protenix v2 biomolecular structure prediction by ByteDance. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Open-source structure prediction neural network for proteins, nucleic acids, and small molecules. State-of-the-art accuracy with multi-chain support.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.