Dock small molecules into proteins using CNN scoring and physics-based pose optimization. Learn more
Dock small molecules into proteins using CNN scoring and physics-based pose optimization. Learn more
Dock small molecules into proteins using CNN scoring and physics-based pose optimization. Learn more
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
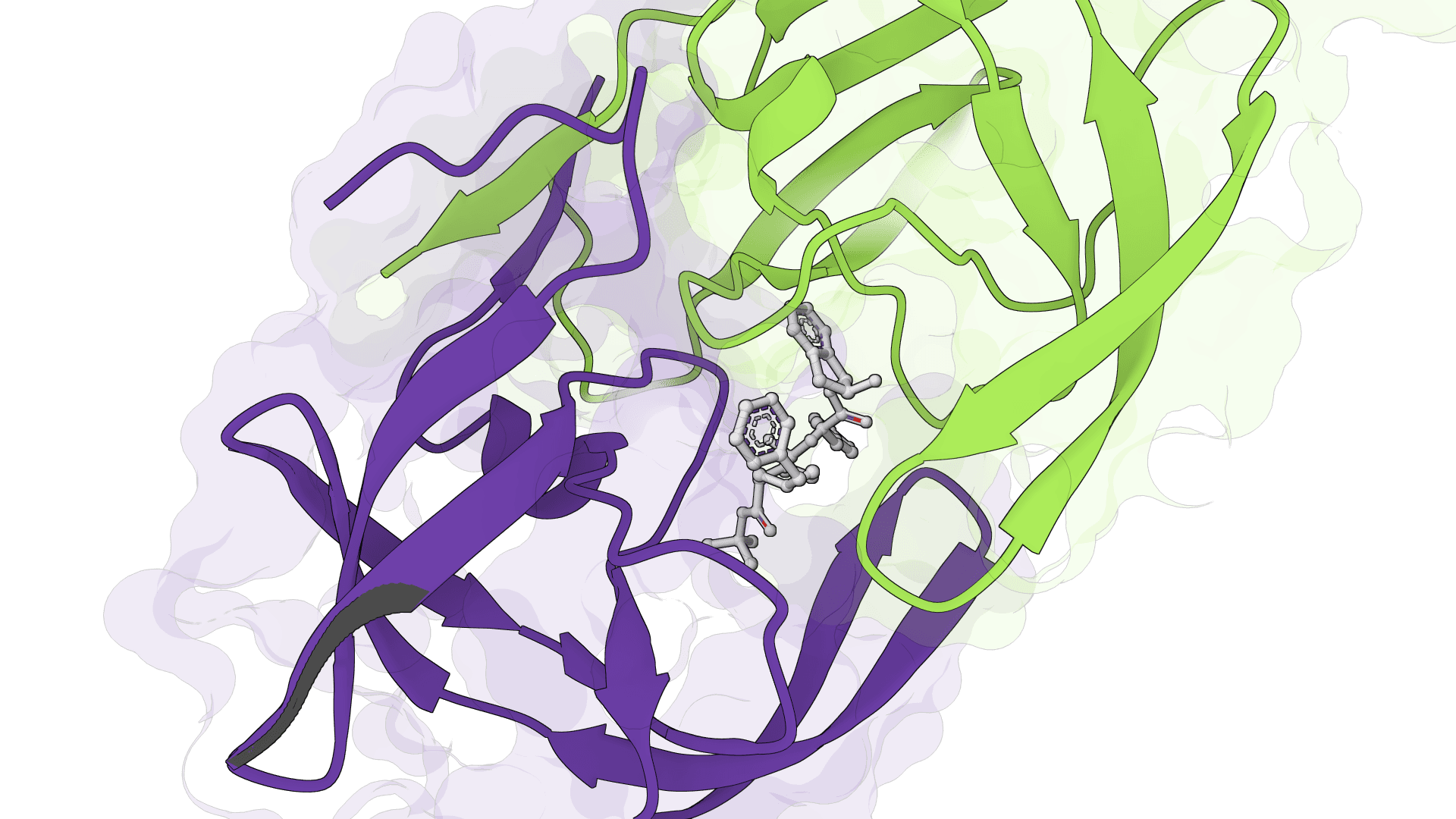
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
(Default CNN ranking) T4 lysozyme L99A—Benzene
(CNN affinity ranking) BRD4 BD1—JQ1
(Empirical-only scoring) Carbonic anhydrase II—Acetazolamide
GNINA (pronounced "NEE-na") docks small molecules into protein structures and ranks the resulting poses with both empirical energy functions and three-dimensional convolutional neural networks. Version 1.3.3 combines the sampling pipeline inherited from AutoDock Vina and Smina with a newer three-model CNN ensemble.
The method is especially useful when the binding site is known and pose selection matters. In the GNINA 1.0 study, CNN rescoring improved top-ranked pose recovery over Vina scoring on both redocking and cross-docking benchmarks. Those benchmark percentages describe curated test sets, not a guaranteed success rate for a new target.
GNINA treats the receptor as rigid in this ProteinIQ workflow. It does not model large induced-fit rearrangements, covalent binding, or explicit flexible side chains. Protonation, tautomer state, retained waters, cofactors, and metals can therefore influence the result as much as the selected docking settings.
Run GNINA online by supplying one protein structure, one small-molecule ligand, and, by default, a positioned reference ligand that marks the binding site. ProteinIQ runs GNINA 1.3.3 on GPU infrastructure and returns ranked SDF poses, CNN and empirical scores, an interactive structure view, a CSV summary, and the complete GNINA log.
| Input | Accepted forms | Requirements |
|---|---|---|
Protein | PDB or ENT file, or RCSB PDB ID | One receptor per job, up to 50 MB. Protein atoms must be present. |
Ligand | SMILES text, SDF, MOL, or MOL2 file, or PubChem lookup | One molecule per job, up to 150 heavy atoms and 300 total atoms. File uploads are limited to 10 MB. |
Reference ligand | Positioned SDF, MOL, or PDB structure | Required while Focus search with reference ligand is enabled. Coordinates must locate the intended pocket in the receptor frame. |
A SMILES ligand is embedded into a three-dimensional starting structure before docking. Query atoms are rejected, and suspected valence problems are reported before submission. Multi-record ligand files are also rejected because each job has one ligand and one ranked pose series.
The reference ligand defines a box only. It does not need to be the compound being docked, but it must occupy the relevant site in the same coordinate system as the receptor. A cognate ligand from a co-crystal structure is usually the clearest choice.
Protein preparation should preserve chemically important metals, cofactors, and waters while removing unrelated crystallization components. PDBFixer can repair missing atoms and common structure-file problems. Ligand fixer can diagnose or repair ligand files that fail chemical parsing.
| Setting | Range and default | Effect |
|---|---|---|
Focus search with reference ligand | On by default | Builds a focused box around the positioned reference ligand. When off, GNINA autoboxes the entire receptor. |
Search box padding | 2 to 12 Å, default 4 Å | Adds space on every side of the autobox. More padding accommodates movement but expands the search volume. |
Exhaustiveness | 1 to 64, default 8 | Sets the number of Monte Carlo search chains. Runtime grows roughly with exhaustiveness. |
Number of poses | 1 to 20, default 9 | Sets the maximum number of ranked poses returned. Diversity filtering can produce fewer poses. |
Min RMSD filter | 0.5 to 3.0 Å, default 1.0 Å | Removes redundant final poses whose pairwise RMSD falls below the threshold. |
Random seed | Optional integer | Repeats the same stochastic search conditions when the receptor, ligand, and settings are unchanged. |
Focused docking with Exhaustiveness 8 is a sensible first pass. Whole-protein autoboxing creates a much larger search space; GNINA documents Exhaustiveness 64 for that workflow. A whole-protein result should still be treated as exploratory because sampling every plausible pocket is substantially harder than searching a known site.
Number of poses is a maximum, not a promise. Raising it does not deepen the search by itself. Exhaustiveness controls sampling effort, while Min RMSD filter controls how similar two retained answers may be.
| Setting | Options and default | Effect |
|---|---|---|
Scoring function | Vina by default; Vinardo, AutoDock4, Dkoes, or Dkoes Fast | Selects the empirical function used for energy evaluation and empirical-guided optimization. |
CNN model | Default ensemble, Fast, and selected Dense, CrossDock, General, or Redock models | Selects the neural network or ensemble used for CNN scoring. |
CNN scoring mode | Rescore by default; Refinement, None, or Full CNN | Determines which docking stages use the CNN. |
Pose sort order | CNN Score by default; CNN Affinity or Empirical Energy | Chooses the value that determines final rank. |
CNN rotations | 0 to 24, default 0 | Averages CNN evaluation across additional rotated views. More rotations increase compute time. |
The default GNINA 1.3 ensemble averages dense_1_3, dense_1_3_PT_KD_3, and crossdock_default2018_KD_4. It is the general choice when pose quality matters. The knowledge-distilled models reduce inference cost and suit larger screening campaigns, while the older named models are useful mainly for reproducing earlier work or testing model sensitivity.
The CNN scoring modes change both runtime and the role of the neural network:
Rescore: Uses the empirical function during sampling and refinement, then applies the CNN to the final poses. This is the standard balance of accuracy and runtime.Refinement: Uses the CNN for final pose refinement and ranking. GNINA reports roughly ten times the runtime of rescoring on GPU, with no universal accuracy gain.None: Uses only the selected empirical function. GNINA forces final ordering to Empirical Energy.Full CNN: Uses the CNN throughout sampling, refinement, and ranking. GNINA describes this as roughly one thousand times slower than rescoring and does not recommend it for routine docking.| Result | Meaning |
|---|---|
Rank | Native GNINA output order according to the effective sort setting. |
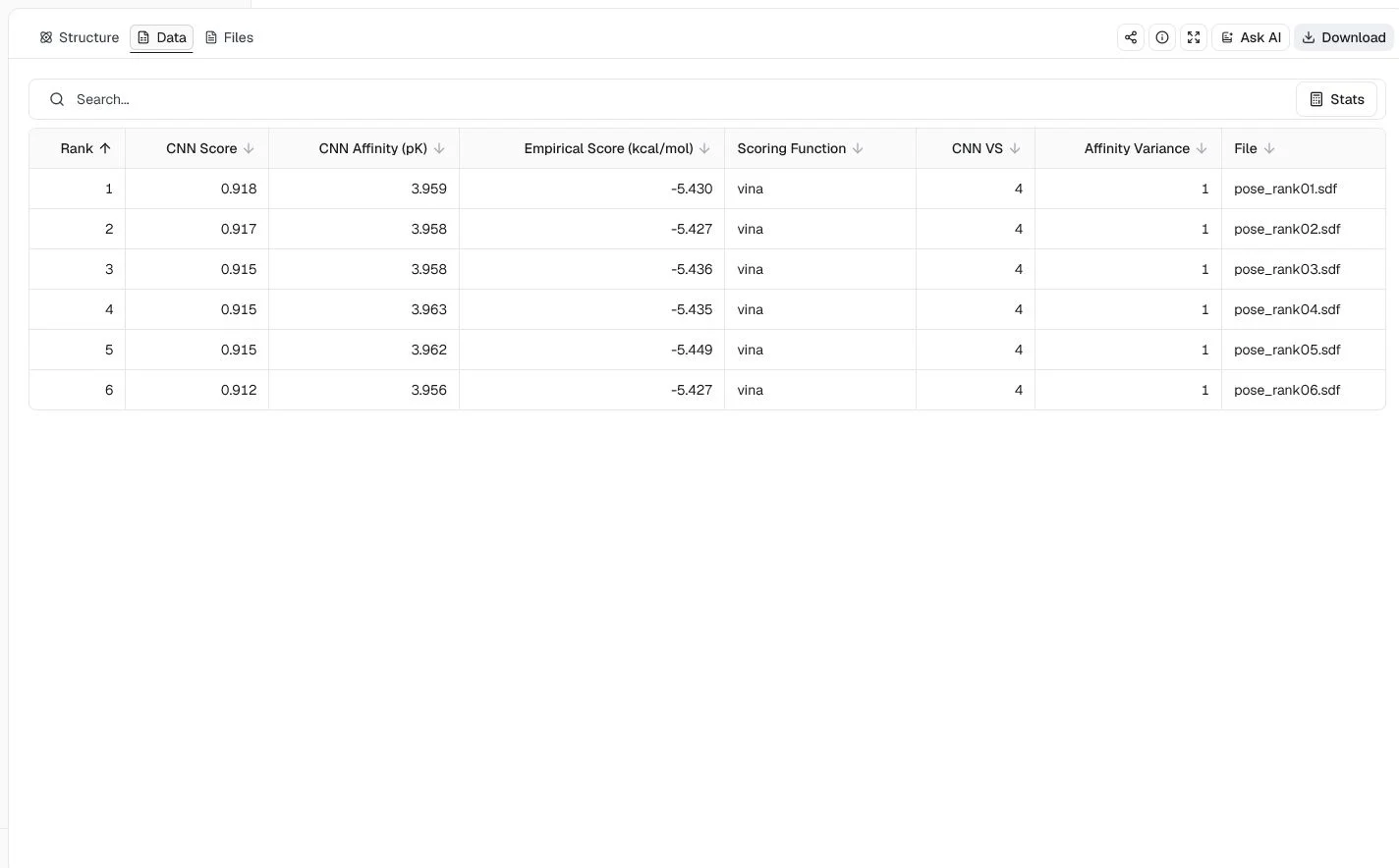
CNN Score | Pose-quality score from 0 to 1. Higher values indicate greater model confidence that the geometry resembles a correct pose. |
CNN Affinity (pK) | Predicted affinity on a pK scale. Higher values indicate stronger predicted binding. |
Empirical Score (kcal/mol) | Minimized value from the selected empirical scoring function. More negative is better within the same function and protocol. |
Scoring Function | The empirical function that produced the empirical score. |
CNN VS | GNINA's virtual-screening composite, calculated as CNN Score × CNN Affinity. Higher values rank more favorably. |
CNN Affinity Variance | Disagreement among models in an ensemble. Lower values mean closer agreement, but GNINA does not provide calibrated good or bad thresholds. |
Minimized RMSD (Å) | Ligand movement during local minimization. It is not RMSD to an experimental binding pose. |
File | Individual lossless SDF record for the ranked pose. |
The file panel includes:
gnina_ranked_poses.sdf: GNINA's complete native ranked SDF outputpose_rankNN.sdf: One lossless SDF record per posedocking_results.csv: Tabular scores and filenamesgnina.log: Captured GNINA standard output and error logGNINA first creates a search box from the reference ligand or, for whole-protein docking, from the receptor. It then runs parallel Monte Carlo chains. Each search step changes ligand translation, rotation, or torsions, locally minimizes the candidate, and accepts or rejects the move with a Metropolis criterion.
After sampling, GNINA pools the best conformations, refines them, removes redundant poses using Min RMSD filter, and calculates the requested scores. The selected Pose sort order determines which value places a pose first.
The CNN receives atom-type density grids centered on the protein-ligand complex. Its two prediction heads answer different questions:
CNN Score: Does this geometry resemble a low-RMSD binding pose?CNN Affinity: How strong is binding predicted to be on the model's learned pK scale?These answers need not agree. A pose can have a plausible predicted affinity but low pose confidence, or high pose confidence without an exceptional affinity estimate. CNN rescoring improves rank selection only among conformations that the search generated; it cannot recover a binding mode that was never sampled.
Start with the top few CNN Score poses and inspect their contacts in the structure viewer. A useful pose should fit inside the intended pocket without severe clashes, preserve key interactions expected for the target, and place chemically compatible groups in sensible environments. A high score does not validate an incorrect protonation state or missing metal coordination.
The GNINA 1.0 benchmark gives useful context for CNN Score, but not a universal cutoff. In that study, a score above 0.8 corresponded to a pose within 2 Å of the crystal pose for at least 79% of redocking cases and 56% of cross-docking cases. Cross-docking is harder because the receptor conformation was solved with a different ligand.
CNN Affinity is expressed as pK, where a one-unit increase represents a tenfold change on the nominal concentration scale. A prediction of 8 is nominally nanomolar-scale, while 6 is micromolar-scale. It is still a learned ranking estimate, not a measured dissociation constant or inhibition constant.
Empirical Score is most useful for comparing poses from runs that use the same empirical function, receptor preparation, search box, and ligand preparation. Vina, Vinardo, AutoDock4, and Dkoes values are not interchangeable. Small score differences should not be treated as precise free-energy differences.
For compound ranking, compare CNN Affinity, CNN VS, and empirical energy rather than selecting on one column alone. Repeated runs with fixed preparation and protocol are more defensible than mixing scores from different box definitions or scoring functions.
CNN Affinity Variance measures model disagreement within an ensemble. It can flag predictions that deserve extra scrutiny, but it has not been calibrated into reliable confidence intervals. A low variance does not prove correctness, and a single-model run cannot provide ensemble disagreement in the same sense.
Minimized RMSD measures how far a pose moved during minimization. It does not compare the docked pose with the reference ligand or a crystal pose. Experimental pose RMSD requires atom mapping to a known ligand conformation and a separate structural comparison.
These completed runs use co-crystal ligand coordinates to define focused search boxes. Each example includes the receptor, every ranked pose, the score table, and downloadable files.
This compact redocking example uses the 4W52 T4 lysozyme L99A structure, benzene, and the co-crystal benzene coordinates. It keeps the default CNN ensemble and CNN Score ordering, requests six poses, and fixes the random seed at 20260724.
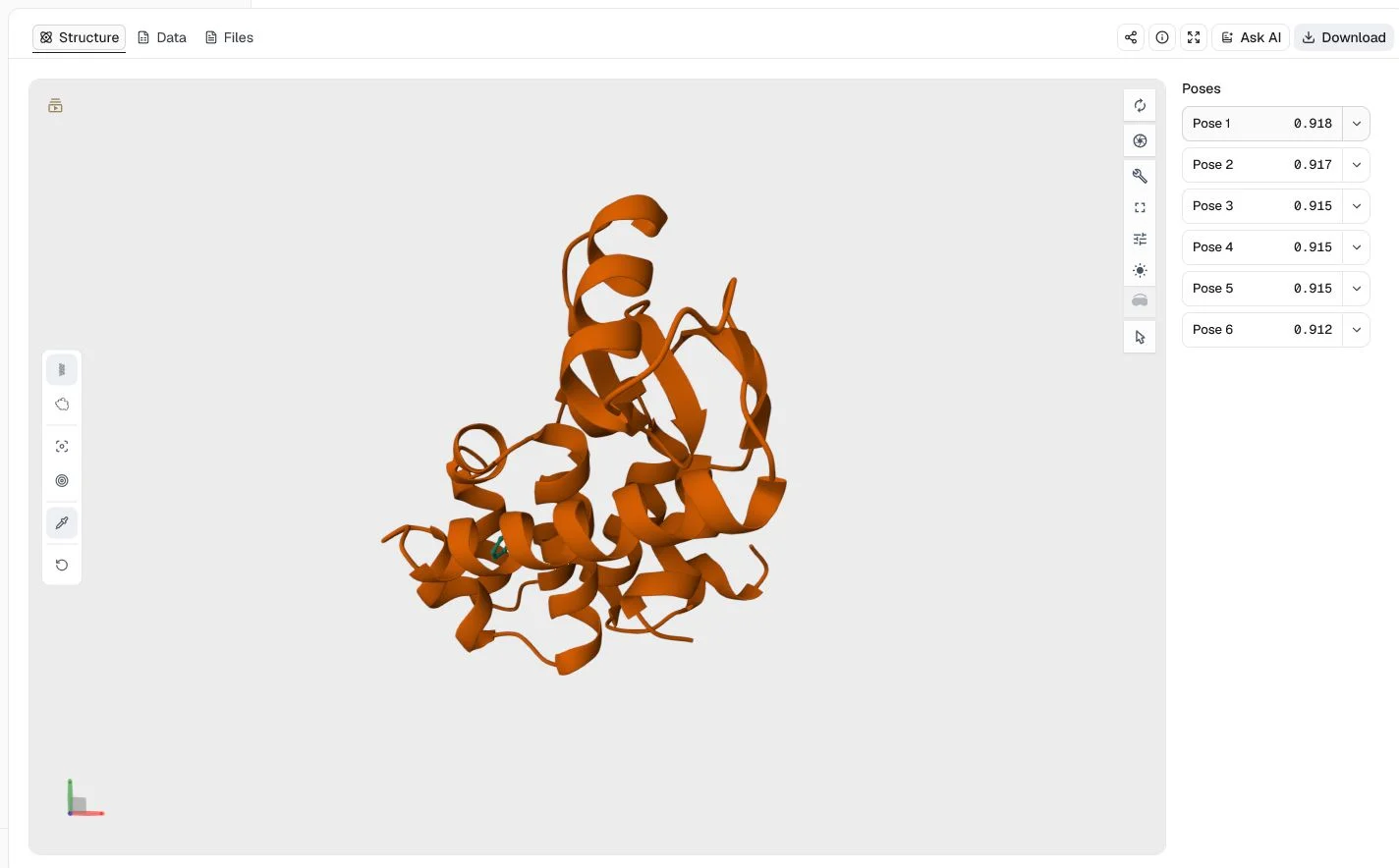
The top pose has a CNN Score of 0.918, while all six poses score above 0.91. The tight cluster shows how GNINA presents closely related high-confidence poses, but it does not establish experimental binding affinity.
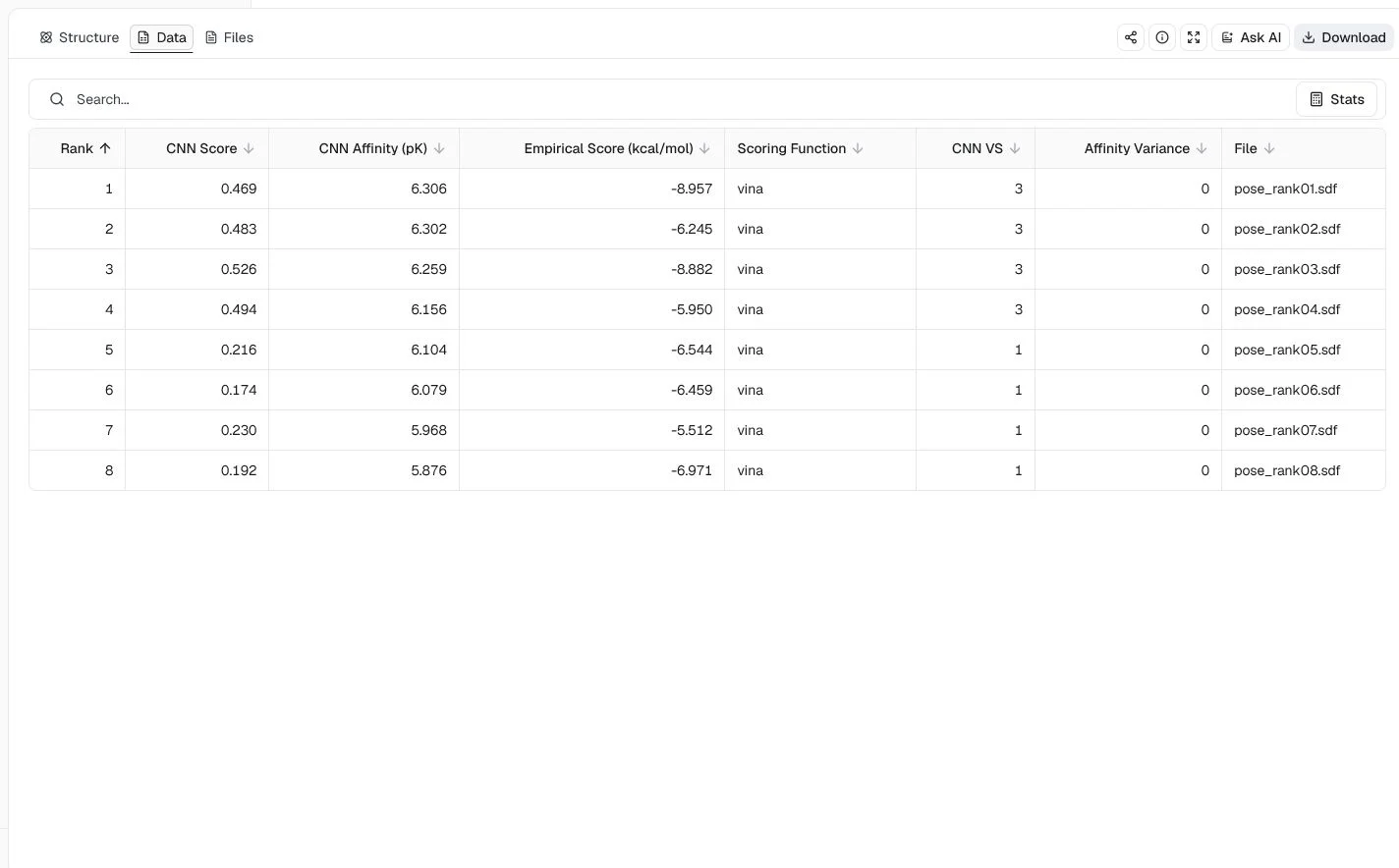
This run uses the 3MXF BRD4 bromodomain structure, JQ1, and the co-crystal JQ1 coordinates. It selects the dense_1_3_PT_KD_3 knowledge-distilled model, performs four CNN rotations, returns eight poses, sorts by CNN Affinity, and fixes the seed at 20260724.
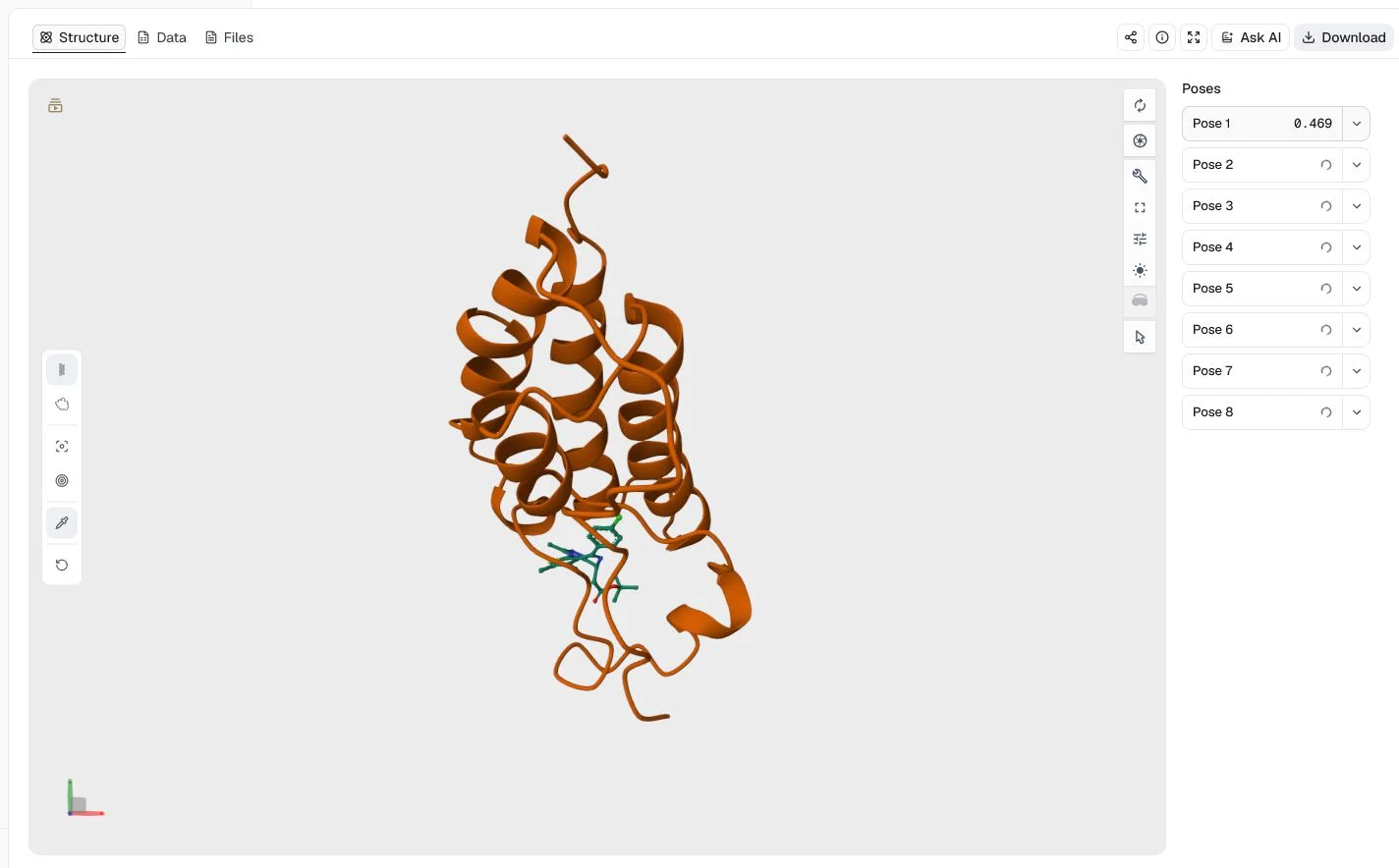
The highest predicted CNN Affinity is 6.306 pK. Because predicted affinity determines the order, the first row does not have the highest CNN Score. Both columns and the binding geometry matter when selecting a pose.
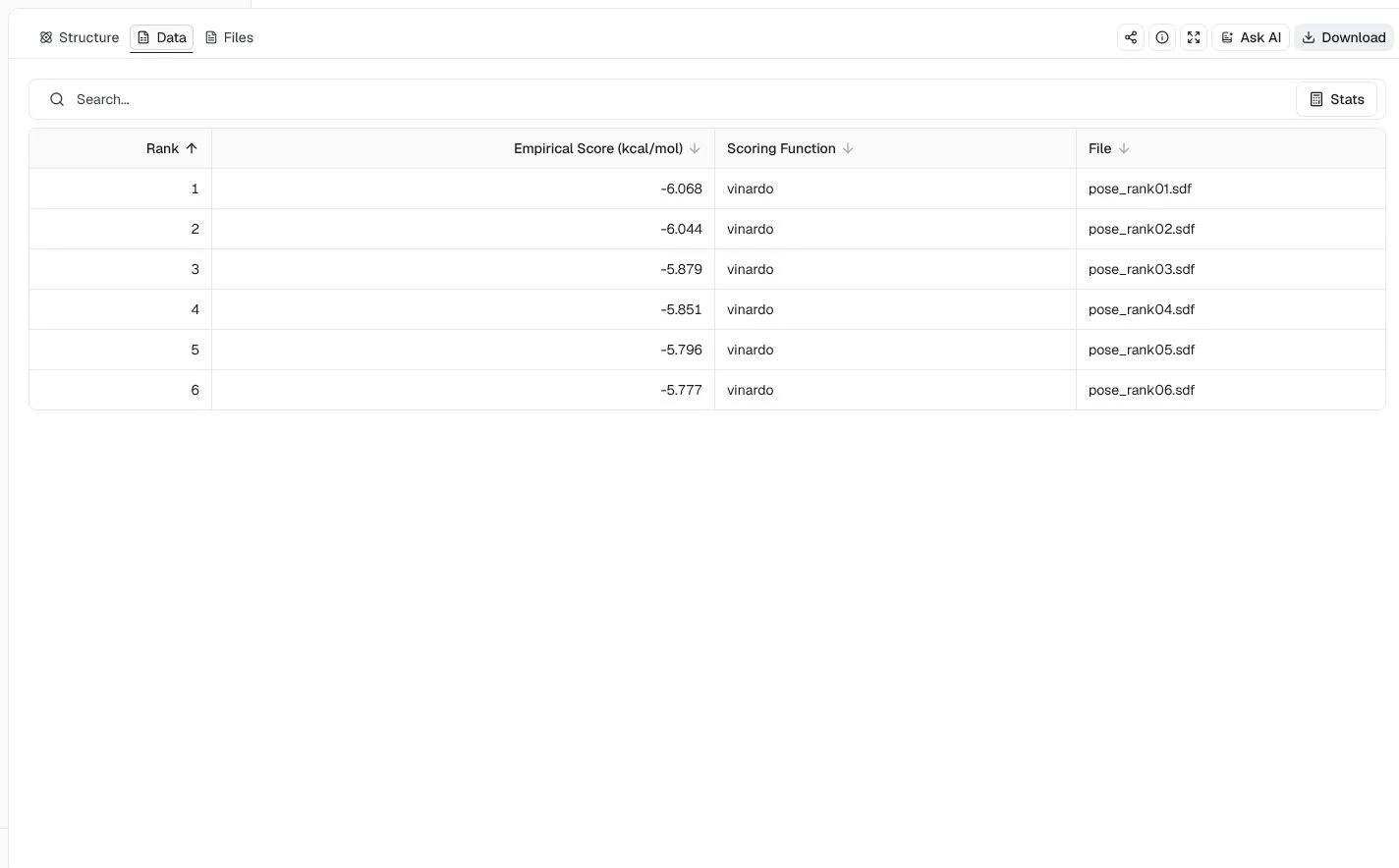
This example retains the zinc ion in the 3HS4 carbonic anhydrase II structure and uses co-crystal acetazolamide coordinates to focus the search. It selects Vinardo, disables CNN scoring, orders six poses by empirical energy, and uses seed 20260724.

The results intentionally contain only Vinardo empirical scores. The best minimized score is −6.068 kcal/mol. Metal coordination, protonation, and retained waters need review before drawing chemical conclusions.
| Method | Search behavior | Scoring | Best fit |
|---|---|---|---|
| GNINA | Vina-family stochastic docking in a defined or receptor-wide box | Empirical energy plus 3D CNN scores | Known-site docking where CNN pose ranking is valuable |
| AutoDock Vina | Stochastic docking in a defined box | Vina empirical score | Fast classical docking and comparisons with established Vina protocols |
| Smina | Vina-family docking with customizable scoring | Empirical and custom scoring functions | Custom scoring terms or Smina protocol reproduction |
| DiffDock | Diffusion-based pose generation without a user-defined pocket | Learned confidence | Blind pose generation when the binding site is unknown |
| SigmaDock | Diffusion-based docking and confidence ranking | Learned confidence | An alternative blind-docking hypothesis generator |
GNINA is a strong default for focused docking, but agreement across methods can be more informative than a single top rank. When no binding site is known, a blind method can generate pocket hypotheses before focused GNINA refinement. When reproducible empirical scoring is the priority, Vina or Smina provides a simpler baseline.