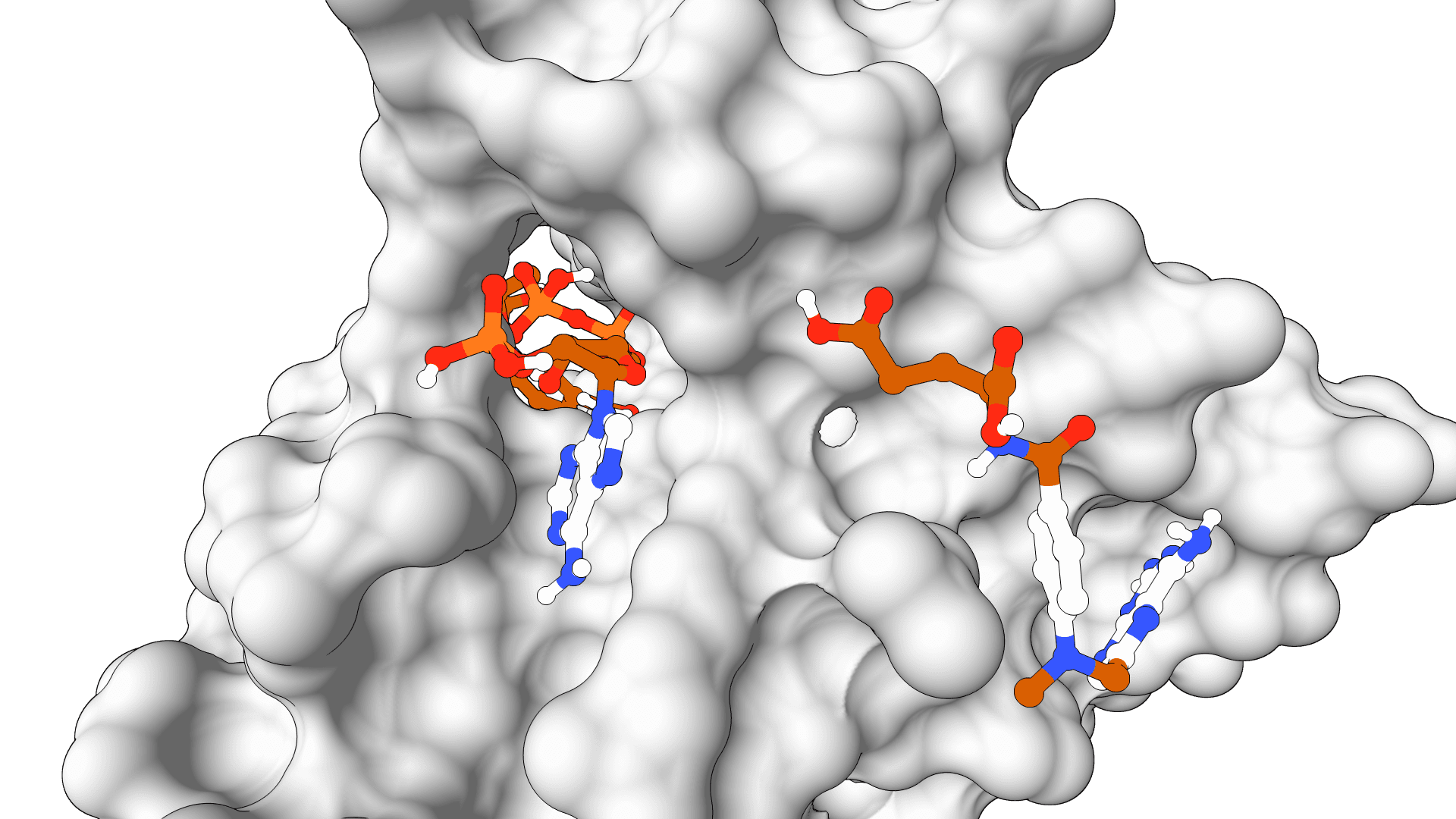
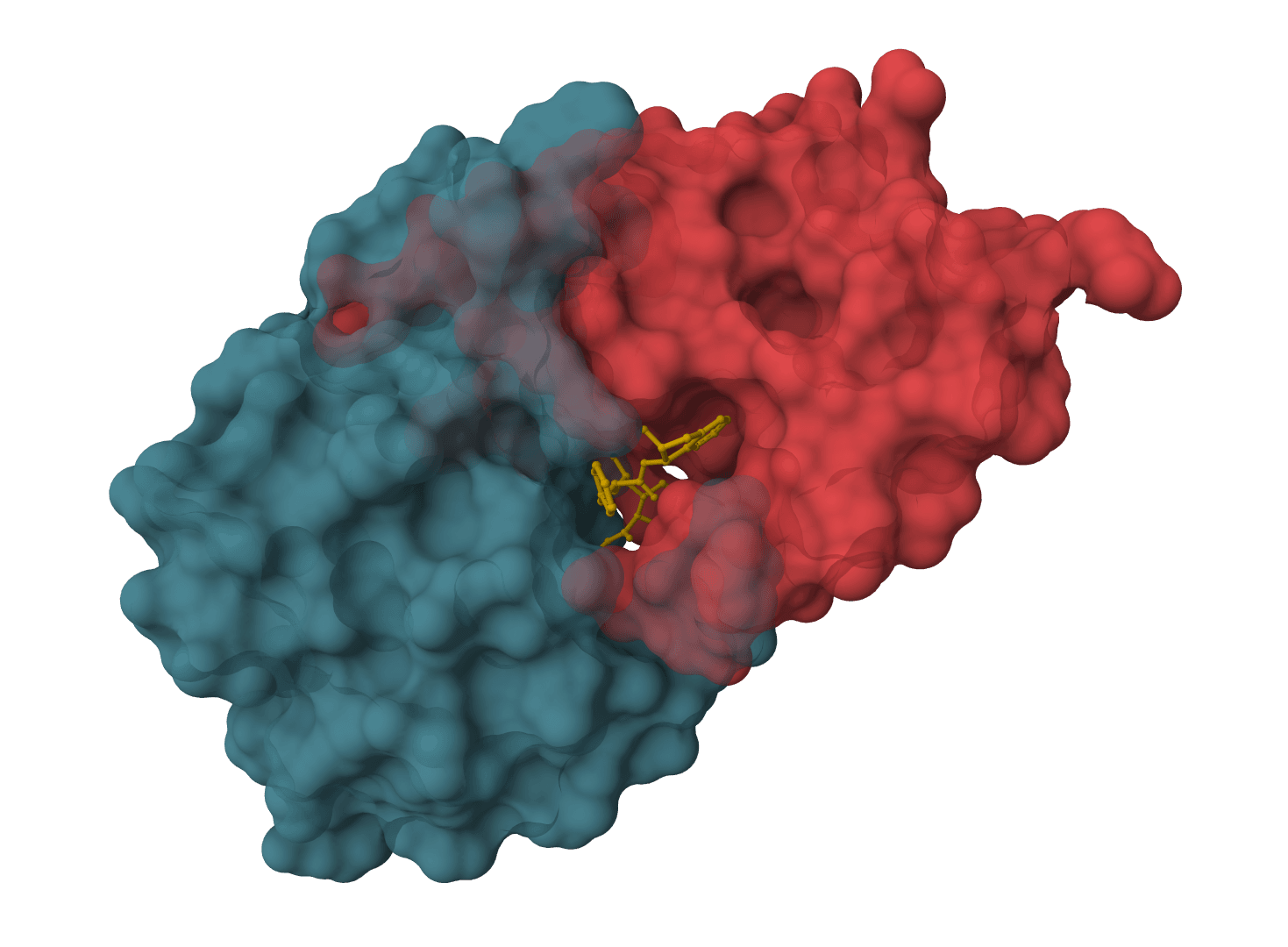
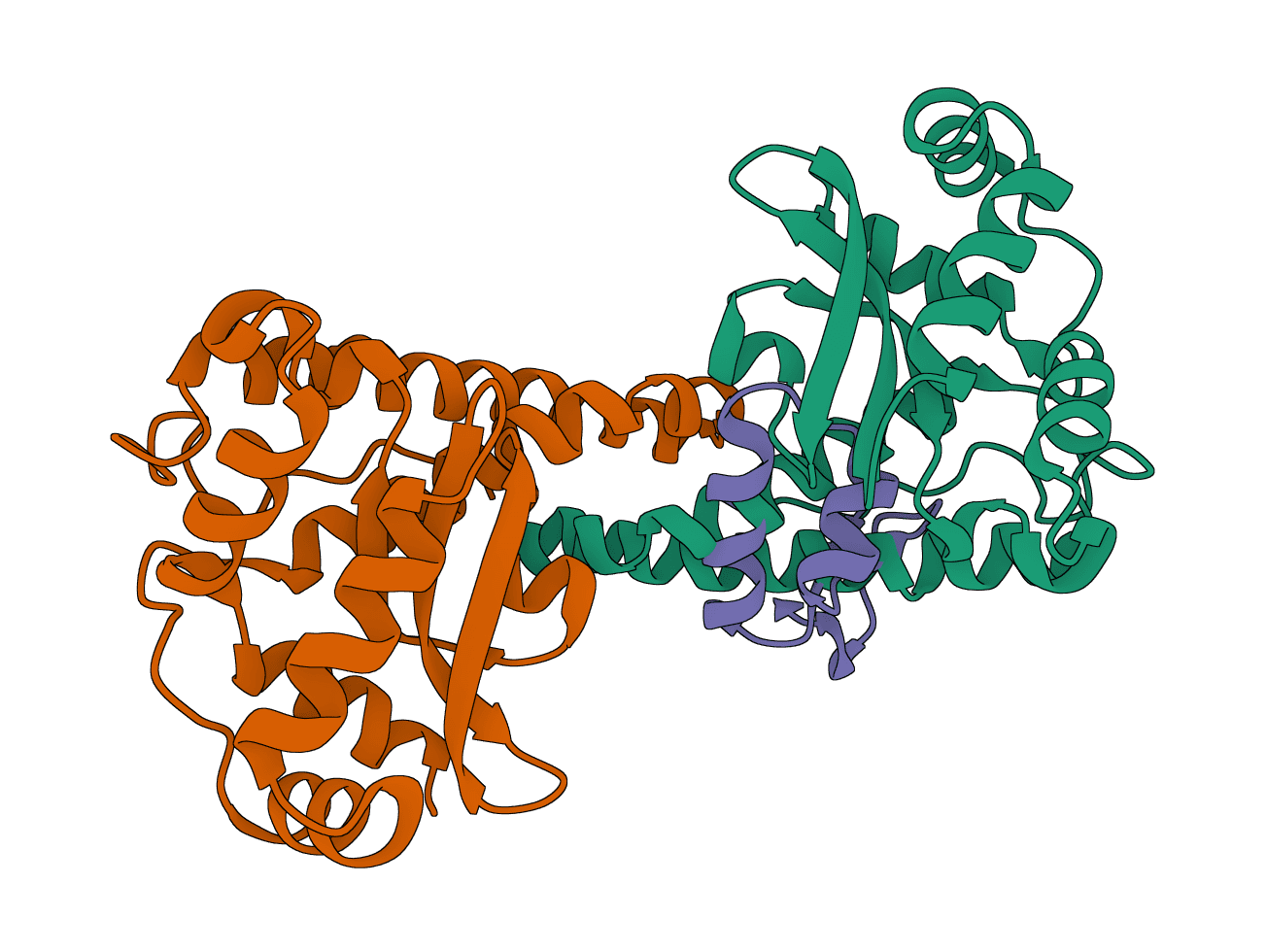LightDock predicts how two macromolecules can form a complex when the binding site is unknown or only partly constrained. It is used for protein-protein, protein-peptide, and protein-DNA docking, including exploratory interface searches and antibody-antigen modeling.
The method comes from the Barcelona Supercomputing Center and is built around Glowworm Swarm Optimization (GSO). Instead of sampling one global search space, LightDock places many local swarms around the receptor surface. Each swarm explores a different region, then the most favorable poses are ranked with the selected scoring function.
LightDock is strongest when broad surface exploration matters. It can also use residue restraints, membrane filters, and Anisotropic Network Model (ANM) modes for limited backbone flexibility, but it is still a docking method for near-rigid structures. Large domain rearrangements, disordered binding partners, and missing interface conformations remain difficult.
Run LightDock online by uploading receptor and ligand structures, or by fetching them from RCSB PDB, then choosing the swarm search, scoring, flexibility, and restraint settings. ProteinIQ returns ranked complex PDB files, LightDock ranking files, the parsed receptor and ligand structures, and pose-level metadata for scoring, clashes, RMSD, swarm, and glowworm identifiers.
| Input | Accepted format | Description |
|---|---|---|
Receptor (larger protein) | .pdb, .ent, or PDB ID | First docking partner. In most protein-protein runs this should be the larger or less mobile structure. Maximum file size is 50 MB. |
Ligand (smaller protein/peptide) | .pdb, .ent, or PDB ID | Second docking partner moved during docking. Protein, peptide, and DNA partners are supported when the selected scoring function is appropriate. Maximum file size is 50 MB. |
Job name | Text | Optional label used to identify the run in job history. |
LightDock works best with cleaned coordinate files. Missing residues, nonstandard atom naming, alternate conformations, and unresolved chains can create failed setup steps or misleading poses. For structure repair before docking, use PDB Fixer.
| Setting | Default | Description |
|---|---|---|
Number of swarms | 0 | Search-space divisions around the receptor. 0 lets LightDock estimate the number of swarms from receptor solvent-accessible surface area. Use a fixed value only when a run needs controlled sampling. |
Glowworms per swarm | 200 | Number of search agents in each swarm. Higher values increase local sampling depth and memory use. |
Simulation steps | 100 | GSO optimization iterations. More steps give each swarm more time to converge, with longer runtime. |
Number of poses to return | 10 | Top ranked complex PDB files returned after global ranking. Range 1 to 50. |
Scoring function | FastDFIRE | Score used during simulation and ranking. FastDFIRE is the fast general choice; DFIRE and DFIRE2 are useful for more accuracy-focused protein docking; dna and ddna are intended for nucleic acid systems. |
Weighted scoring configuration | Empty | Optional LightDock multi-scoring configuration. Leave empty to use the selected scoring function directly. |
| Setting | Default | Description |
|---|---|---|
Receptor flexibility (ANM modes) | 0 | Number of receptor ANM modes. 0 runs rigid docking. Values from 5 to 10 add backbone deformation along low-frequency normal modes. |
Ligand flexibility (ANM modes) | 0 | Number of ligand ANM modes. This is often more useful for peptides or small flexible protein domains than for rigid globular partners. |
Ignore hydrogens | false | Removes hydrogen atoms before setup. This is commonly enabled with FastDFIRE because many hydrogen atoms are not parameterized by that scoring function. |
Ignore OXT atoms | false | Removes terminal OXT atoms. Useful when a scoring function or input preparation step does not handle terminal oxygens consistently. |
Ignore water | false | Removes water molecules. Enable when crystallographic waters are not part of the intended docking model. |
Local minimization | false | Enables LightDock local minimization during GSO simulation. This can improve local contacts but adds runtime. |
Starting-points seed | 324324 | Random seed for initial swarm placement. |
GSO seed | 324324 | Random seed for the GSO simulation. |
ANM seed | 324324 | Random seed for ANM extent sampling when flexibility is enabled. |
Receptor ANM RMSD | 0.5 | RMSD interval for receptor ANM conformations. |
Ligand ANM RMSD | 0.5 | RMSD interval for ligand ANM conformations. |
Translation step | 0.5 | Normalized translation step used during GSO movement. |
Rotation step | 0.5 | Normalized rotation step used during rotational updates. |
Normal-modes step | 0.5 | Normalized step for ANM mode amplitudes. |
| Setting | Default | Description |
|---|---|---|
Surface density | 50 | Density used when automatic swarm placement estimates how many swarms to create. |
Swarm radius | 10 | Initial local search radius in angstroms. |
Fixed swarm distance | 0 | Optional fixed distance from the receptor surface. 0 lets LightDock estimate distance from ligand size. |
Swarms per restraint | 20 | Maximum swarms kept around each receptor restraint. |
Dense sampling | false | Uses denser restraint-swarm filtering. |
Cluster generated poses | false | Runs BSAS clustering before global ranking. This can be substantially slower because LightDock must generate conformations for all glowworms. |
Clashes cutoff | Empty | Optional clash cutoff applied during LightDock ranking. |
Restraints file | Empty | Optional LightDock restraint content. Lines use R or L for receptor or ligand, then residue identity and an active, passive, or blocked label. Example: R A.ALA.42 A. |
Flip restrained poses | false | Applies LightDock's 180-degree flip to half of the starting poses when restraints are used. |
Membrane filter | false | Enables LightDock membrane restraint filtering. |
Transmembrane filter | false | Enables transmembrane restraint filtering. |
Write starting positions | false | Returns LightDock setup support files for inspecting initial swarm placement. |
LightDock returns one PDB file for each selected complex. Each complex contains the receptor and ligand in the same coordinate frame, with ligand chain IDs reassigned when needed to avoid conflicts with receptor chains.
The spreadsheet is sorted by LightDock's rank_by_scoring.list order. The top row is the best ranked pose for the selected scoring function.
| Column | Description |
|---|---|
Rank | Global rank from rank_by_scoring.list. Rank 1 is the best pose in the returned set. |
Score | Selected LightDock scoring value. The numeric scale depends on the scoring function, so compare scores within the same run and scoring setup. |
Swarm | Search region that produced the pose. Multiple high-ranking poses from the same swarm suggest local convergence around one interface. |
Glowworm | Search agent identifier within the swarm. |
Luciferin | Internal GSO quality value used by glowworm movement. |
Neighbors | Number of neighboring glowworms considered during movement. |
Vision range | Local radius used for glowworm neighbor detection. |
RMSD | RMSD value reported by the LightDock ranking file. Its meaning depends on the available ranking context and should not be read as native accuracy unless a reference was used. |
Clashes | Number of steric clashes reported during ranking. Lower clash counts are preferred when scores are similar. |
File | Downloadable complex PDB file. |
Additional downloadable files include:
rank_by_scoring.list: Global ranking by the selected scoring function.solutions.list: Combined LightDock solutions file.rank_by_luciferin.list: Ranking by GSO luciferin value.rank_by_rmsd.list: Ranking by RMSD value when available.setup.json: LightDock setup configuration.lightdock.info: Simulation metadata.swarm_centers.pdb: Initial swarm centers around the receptor.lightdock_receptor.pdb and lightdock_ligand.pdb: Structures parsed by LightDock during setup.LightDock treats docking as many local optimization problems. The receptor surface is sampled with swarms, and each swarm contains a population of glowworms. A glowworm encodes a possible ligand pose through translation, rotation, and optional ANM deformation parameters.
During simulation, glowworms move toward nearby glowworms with better luciferin values. Since movement is local to each swarm, different regions of the receptor surface can converge on different candidate interfaces. This is useful for blind docking because the method does not need one predefined binding pocket.
The scoring function is part of the search, not only a final rescoring step. FastDFIRE, DFIRE, DFIRE2, pyDock, PISA, shape complementarity, DNA-specific scoring, and several specialized potentials are available. A multi-scoring configuration can combine more than one scoring function, but memory use rises because each glowworm keeps scoring state for each configured function.
ANM flexibility adds low-frequency backbone deformation modes through ProDy. This is not molecular dynamics. It lets receptor and ligand structures move along coarse collective modes, which can help with loop shifts or moderate interface adjustment, but it will not solve binding events that require a different folded state.
The rank order matters more than the raw score. A score from a FastDFIRE run should not be compared directly with a score from a PISA, shape-complementarity, or DNA-specific run. Even within one scoring function, absolute values depend on interface size, atom composition, and preprocessing choices.
A practical first pass is:
5 to 10 poses in the 3D viewer.For blind ab initio docking, LightDock benchmark performance is modest because the search space is large. The original LightDock study reported better results when flexibility was enabled, and the information-driven LightDock work showed much stronger success rates when interface restraints were available. In practice, blind runs are best treated as hypothesis generation rather than final structural evidence.
| Tool | Best fit | Main tradeoff |
|---|---|---|
LightDock | Broad protein-protein, protein-peptide, or protein-DNA surface search with optional restraints and ANM flexibility | Strong exploratory sampling, but results need careful ranking and structural inspection |
| HADDOCK3 | Protein-protein docking with reliable interface residues, mutagenesis data, cross-links, or other restraints | More data-driven and interpretable when restraints exist, but less convenient for wide blind exploration |
| DFMDock | Fast structure-only protein-protein docking with learned rigid-body sampling | Good for quick unrestrained screening, but does not model explicit LightDock-style swarms or ANM flexibility |
| AF2Dock | AlphaFold2-guided protein-protein docking from two input structures | Better when AF2-style refinement and confidence outputs are desired |
| GeoDock | Learned geometry-aware protein complex prediction | Better when a fast geometric deep-learning model is the preferred starting point |
| ColabDock | AlphaFold2-style complex prediction guided by restraints | Useful when the workflow depends on AlphaFold-derived confidence and restraint satisfaction |
| EquiDock | Very fast rigid protein-protein pose prediction | Lightweight and fast, but returns less search diversity |
| DockQ | Evaluating a predicted complex against a known reference complex | Validation tool, not a docking generator |
| PPAP | Predicting affinity for a protein-protein complex | Scoring tool, not a docking generator |
For small-molecule docking, use AutoDock Vina, GNINA, or DiffDock. LightDock is designed for macromolecular partners.
Input extraction failed: Confirm that both receptor and ligand are valid PDB or ENT structures, or valid four-character PDB IDs available from RCSB.LightDock setup failed: Check atom records, chain IDs, residue names, alternate conformations, and missing coordinates. Removing waters, hydrogens, or OXT atoms often helps with FastDFIRE runs.No ranked LightDock poses found: Reduce filtering, remove a strict clash cutoff, or run without clustering. Also confirm that restraints do not exclude all relevant swarms.No complex structures generated: Lower the number of swarms or steps for a quick diagnostic run, then increase sampling once setup succeeds.LightDock execution failed: Retry with defaults: automatic swarms, 200 glowworms, 100 steps, FastDFIRE, no clustering, and no optional filters. Add advanced options one at a time.
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
AF2Dock adapts AlphaFold2-style co-folding for structure-based protein-protein docking. It docks receptor and ligand protein structures with flow-matching refinement and ranks sampled complexes by iPTM.
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
GeoDock predicts flexible protein-protein docking complexes from two separate protein structures using a multi-track iterative transformer and the DIPS 0.3 checkpoint from the Gray Lab release.
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
LightDock predicts how two macromolecules can form a complex when the binding site is unknown or only partly constrained. It is used for protein-protein, protein-peptide, and protein-DNA docking, including exploratory interface searches and antibody-antigen modeling.
The method comes from the Barcelona Supercomputing Center and is built around Glowworm Swarm Optimization (GSO). Instead of sampling one global search space, LightDock places many local swarms around the receptor surface. Each swarm explores a different region, then the most favorable poses are ranked with the selected scoring function.
LightDock is strongest when broad surface exploration matters. It can also use residue restraints, membrane filters, and Anisotropic Network Model (ANM) modes for limited backbone flexibility, but it is still a docking method for near-rigid structures. Large domain rearrangements, disordered binding partners, and missing interface conformations remain difficult.
Run LightDock online by uploading receptor and ligand structures, or by fetching them from RCSB PDB, then choosing the swarm search, scoring, flexibility, and restraint settings. ProteinIQ returns ranked complex PDB files, LightDock ranking files, the parsed receptor and ligand structures, and pose-level metadata for scoring, clashes, RMSD, swarm, and glowworm identifiers.
| Input | Accepted format | Description |
|---|---|---|
Receptor (larger protein) | .pdb, .ent, or PDB ID | First docking partner. In most protein-protein runs this should be the larger or less mobile structure. Maximum file size is 50 MB. |
Ligand (smaller protein/peptide) | .pdb, .ent, or PDB ID | Second docking partner moved during docking. Protein, peptide, and DNA partners are supported when the selected scoring function is appropriate. Maximum file size is 50 MB. |
Job name | Text | Optional label used to identify the run in job history. |
LightDock works best with cleaned coordinate files. Missing residues, nonstandard atom naming, alternate conformations, and unresolved chains can create failed setup steps or misleading poses. For structure repair before docking, use PDB Fixer.
| Setting | Default | Description |
|---|---|---|
Number of swarms | 0 | Search-space divisions around the receptor. 0 lets LightDock estimate the number of swarms from receptor solvent-accessible surface area. Use a fixed value only when a run needs controlled sampling. |
Glowworms per swarm | 200 | Number of search agents in each swarm. Higher values increase local sampling depth and memory use. |
Simulation steps | 100 | GSO optimization iterations. More steps give each swarm more time to converge, with longer runtime. |
Number of poses to return | 10 | Top ranked complex PDB files returned after global ranking. Range 1 to 50. |
Scoring function | FastDFIRE | Score used during simulation and ranking. FastDFIRE is the fast general choice; DFIRE and DFIRE2 are useful for more accuracy-focused protein docking; dna and ddna are intended for nucleic acid systems. |
Weighted scoring configuration | Empty | Optional LightDock multi-scoring configuration. Leave empty to use the selected scoring function directly. |
| Setting | Default | Description |
|---|---|---|
Receptor flexibility (ANM modes) | 0 | Number of receptor ANM modes. 0 runs rigid docking. Values from 5 to 10 add backbone deformation along low-frequency normal modes. |
Ligand flexibility (ANM modes) | 0 | Number of ligand ANM modes. This is often more useful for peptides or small flexible protein domains than for rigid globular partners. |
Ignore hydrogens | false | Removes hydrogen atoms before setup. This is commonly enabled with FastDFIRE because many hydrogen atoms are not parameterized by that scoring function. |
Ignore OXT atoms | false | Removes terminal OXT atoms. Useful when a scoring function or input preparation step does not handle terminal oxygens consistently. |
Ignore water | false | Removes water molecules. Enable when crystallographic waters are not part of the intended docking model. |
Local minimization | false | Enables LightDock local minimization during GSO simulation. This can improve local contacts but adds runtime. |
Starting-points seed | 324324 | Random seed for initial swarm placement. |
GSO seed | 324324 | Random seed for the GSO simulation. |
ANM seed | 324324 | Random seed for ANM extent sampling when flexibility is enabled. |
Receptor ANM RMSD | 0.5 | RMSD interval for receptor ANM conformations. |
Ligand ANM RMSD | 0.5 | RMSD interval for ligand ANM conformations. |
Translation step | 0.5 | Normalized translation step used during GSO movement. |
Rotation step | 0.5 | Normalized rotation step used during rotational updates. |
Normal-modes step | 0.5 | Normalized step for ANM mode amplitudes. |
| Setting | Default | Description |
|---|---|---|
Surface density | 50 | Density used when automatic swarm placement estimates how many swarms to create. |
Swarm radius | 10 | Initial local search radius in angstroms. |
Fixed swarm distance | 0 | Optional fixed distance from the receptor surface. 0 lets LightDock estimate distance from ligand size. |
Swarms per restraint | 20 | Maximum swarms kept around each receptor restraint. |
Dense sampling | false | Uses denser restraint-swarm filtering. |
Cluster generated poses | false | Runs BSAS clustering before global ranking. This can be substantially slower because LightDock must generate conformations for all glowworms. |
Clashes cutoff | Empty | Optional clash cutoff applied during LightDock ranking. |
Restraints file | Empty | Optional LightDock restraint content. Lines use R or L for receptor or ligand, then residue identity and an active, passive, or blocked label. Example: R A.ALA.42 A. |
Flip restrained poses | false | Applies LightDock's 180-degree flip to half of the starting poses when restraints are used. |
Membrane filter | false | Enables LightDock membrane restraint filtering. |
Transmembrane filter | false | Enables transmembrane restraint filtering. |
Write starting positions | false | Returns LightDock setup support files for inspecting initial swarm placement. |
LightDock returns one PDB file for each selected complex. Each complex contains the receptor and ligand in the same coordinate frame, with ligand chain IDs reassigned when needed to avoid conflicts with receptor chains.
The spreadsheet is sorted by LightDock's rank_by_scoring.list order. The top row is the best ranked pose for the selected scoring function.
| Column | Description |
|---|---|
Rank | Global rank from rank_by_scoring.list. Rank 1 is the best pose in the returned set. |
Score | Selected LightDock scoring value. The numeric scale depends on the scoring function, so compare scores within the same run and scoring setup. |
Swarm | Search region that produced the pose. Multiple high-ranking poses from the same swarm suggest local convergence around one interface. |
Glowworm | Search agent identifier within the swarm. |
Luciferin | Internal GSO quality value used by glowworm movement. |
Neighbors | Number of neighboring glowworms considered during movement. |
Vision range | Local radius used for glowworm neighbor detection. |
RMSD | RMSD value reported by the LightDock ranking file. Its meaning depends on the available ranking context and should not be read as native accuracy unless a reference was used. |
Clashes | Number of steric clashes reported during ranking. Lower clash counts are preferred when scores are similar. |
File | Downloadable complex PDB file. |
Additional downloadable files include:
rank_by_scoring.list: Global ranking by the selected scoring function.solutions.list: Combined LightDock solutions file.rank_by_luciferin.list: Ranking by GSO luciferin value.rank_by_rmsd.list: Ranking by RMSD value when available.setup.json: LightDock setup configuration.lightdock.info: Simulation metadata.swarm_centers.pdb: Initial swarm centers around the receptor.lightdock_receptor.pdb and lightdock_ligand.pdb: Structures parsed by LightDock during setup.LightDock treats docking as many local optimization problems. The receptor surface is sampled with swarms, and each swarm contains a population of glowworms. A glowworm encodes a possible ligand pose through translation, rotation, and optional ANM deformation parameters.
During simulation, glowworms move toward nearby glowworms with better luciferin values. Since movement is local to each swarm, different regions of the receptor surface can converge on different candidate interfaces. This is useful for blind docking because the method does not need one predefined binding pocket.
The scoring function is part of the search, not only a final rescoring step. FastDFIRE, DFIRE, DFIRE2, pyDock, PISA, shape complementarity, DNA-specific scoring, and several specialized potentials are available. A multi-scoring configuration can combine more than one scoring function, but memory use rises because each glowworm keeps scoring state for each configured function.
ANM flexibility adds low-frequency backbone deformation modes through ProDy. This is not molecular dynamics. It lets receptor and ligand structures move along coarse collective modes, which can help with loop shifts or moderate interface adjustment, but it will not solve binding events that require a different folded state.
The rank order matters more than the raw score. A score from a FastDFIRE run should not be compared directly with a score from a PISA, shape-complementarity, or DNA-specific run. Even within one scoring function, absolute values depend on interface size, atom composition, and preprocessing choices.
A practical first pass is:
5 to 10 poses in the 3D viewer.For blind ab initio docking, LightDock benchmark performance is modest because the search space is large. The original LightDock study reported better results when flexibility was enabled, and the information-driven LightDock work showed much stronger success rates when interface restraints were available. In practice, blind runs are best treated as hypothesis generation rather than final structural evidence.
| Tool | Best fit | Main tradeoff |
|---|---|---|
LightDock | Broad protein-protein, protein-peptide, or protein-DNA surface search with optional restraints and ANM flexibility | Strong exploratory sampling, but results need careful ranking and structural inspection |
| HADDOCK3 | Protein-protein docking with reliable interface residues, mutagenesis data, cross-links, or other restraints | More data-driven and interpretable when restraints exist, but less convenient for wide blind exploration |
| DFMDock | Fast structure-only protein-protein docking with learned rigid-body sampling | Good for quick unrestrained screening, but does not model explicit LightDock-style swarms or ANM flexibility |
| AF2Dock | AlphaFold2-guided protein-protein docking from two input structures | Better when AF2-style refinement and confidence outputs are desired |
| GeoDock | Learned geometry-aware protein complex prediction | Better when a fast geometric deep-learning model is the preferred starting point |
| ColabDock | AlphaFold2-style complex prediction guided by restraints | Useful when the workflow depends on AlphaFold-derived confidence and restraint satisfaction |
| EquiDock | Very fast rigid protein-protein pose prediction | Lightweight and fast, but returns less search diversity |
| DockQ | Evaluating a predicted complex against a known reference complex | Validation tool, not a docking generator |
| PPAP | Predicting affinity for a protein-protein complex | Scoring tool, not a docking generator |
For small-molecule docking, use AutoDock Vina, GNINA, or DiffDock. LightDock is designed for macromolecular partners.
Input extraction failed: Confirm that both receptor and ligand are valid PDB or ENT structures, or valid four-character PDB IDs available from RCSB.LightDock setup failed: Check atom records, chain IDs, residue names, alternate conformations, and missing coordinates. Removing waters, hydrogens, or OXT atoms often helps with FastDFIRE runs.No ranked LightDock poses found: Reduce filtering, remove a strict clash cutoff, or run without clustering. Also confirm that restraints do not exclude all relevant swarms.No complex structures generated: Lower the number of swarms or steps for a quick diagnostic run, then increase sampling once setup succeeds.LightDock execution failed: Retry with defaults: automatic swarms, 200 glowworms, 100 steps, FastDFIRE, no clustering, and no optional filters. Add advanced options one at a time.HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
AF2Dock adapts AlphaFold2-style co-folding for structure-based protein-protein docking. It docks receptor and ligand protein structures with flow-matching refinement and ranks sampled complexes by iPTM.
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
GeoDock predicts flexible protein-protein docking complexes from two separate protein structures using a multi-track iterative transformer and the DIPS 0.3 checkpoint from the Gray Lab release.
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.