Dock ligands into protein structures and estimate binding modes, poses, and affinity scores. Learn more
Dock ligands into protein structures and estimate binding modes, poses, and affinity scores. Learn more
Dock ligands into protein structures and estimate binding modes, poses, and affinity scores. Learn more
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
(Single mode) c-Abl kinase—Imatinib
(Simultaneous co-docking) Human DHFR—Methotrexate + NADPH
(Batch mode) c-Abl kinase—Inhibitor panel
AutoDock Vina is an open-source molecular docking engine for protein-ligand complexes. It searches possible small-molecule poses inside a defined region of a receptor and ranks them with an empirical scoring function. Originally developed by Oleg Trott in the Molecular Graphics Laboratory, Vina is now maintained by the Forli Lab at Scripps Research.
The result is a binding hypothesis, not a measured affinity or proof of a binding mode. Vina is most useful when the binding region is known and the receptor, ligand, and search protocol can be prepared consistently.
ProteinIQ supports Vina, Vinardo, and map-based AutoDock4 scoring, plus flexible residues, AutoDock4Zn preparation, hydrated docking, simultaneous co-docking, and independent batch runs. For larger virtual screens, consider AutoDock GPU.
On ProteinIQ, AutoDock Vina is most commonly used to:
Run AutoDock Vina online by supplying a receptor structure and one or more small-molecule ligands, then defining the binding region from a bound ligand, selected residues, manual coordinates, or the whole protein. ProteinIQ runs Vina 1.2.7 and returns ranked PDBQT poses, affinity estimates, RMSD bounds, prepared structures, and preparation records.
Runtime depends strongly on box volume, ligand flexibility, exhaustiveness, flexible residues, and ligand count.
A typical docking run follows this sequence:
Exhaustiveness of 8, then increase search effort only when repeated runs do not converge on a stable pose family.AutoDock Vina requires two inputs: a protein receptor and a ligand (or multiple ligands with simultaneous co-docking and batch docking).
| Input | Description |
|---|---|
Receptor | Accepts .pdb, .ent, .cif, .mmcif, or an already prepared rigid .pdbqt, or fetches a 4-character PDB ID such as 1HSG. Files may be up to 50 MB. Source structures use PDB2PQR 3.7.1 by default, followed by Meeko 0.7.1. PDB2PQR can be disabled in Advanced settings. A prepared PDBQT is used unchanged and cannot also be split into flexible residues. |
Ligand | Accepts SMILES text, a supported structure file (.pdbqt, .sdf, .mol, .mol2, .smiles, .smi, .txt, .csv), or a PubChem fetch. Files may be up to 50 MB. Each ligand is limited to 32 rotatable bonds, 150 heavy atoms, and 300 total atoms. One ligand is used in Single ligand mode, Simultaneous co-docking accepts up to 5 ligands in one shared search, and Batch docking docks up to 10 ligands independently against the same receptor. |
Ligand mode | Single ligand runs one docking job, Simultaneous co-docking places multiple ligands in the same search space during one run, and Batch docking runs independent ligand jobs for focused virtual screening. |
Job name | Optional label stored with the run to make repeated docking experiments easier to identify. |
Ligand validation blocks metal-containing ligands and disconnected multi-fragment submissions in the standard Vina workflow. Those cases require a docking setup that explicitly supports their chemistry; GNINA is an alternative when CNN pose scoring is useful, but it does not remove the need to validate unusual ligand chemistry.
SMILES, SDF, MOL, and compatible MOL2 ligands follow the same RDKit and Meeko preparation path. MOL2 records with unsupported atom types are normalized through Open Babel before Meeko preparation. An already prepared ligand PDBQT passes through unchanged.
Score only and Local only do not perform a global search. They require a pre-positioned three-dimensional ligand whose coordinates already place it inside the binding site. Dock is the appropriate operation for SMILES, PubChem records, or ligand files that are not already positioned in the receptor coordinate frame.
Text and CSV ligand files are expanded into individual ligands before mode and plan limits are checked:
| Format | Accepted rows |
|---|---|
.csv | A case-insensitive smiles header, with an optional name, compound, or id column. Other headed columns are ignored. Headerless files may contain one SMILES value per row or name,smiles. Standard CSV quoting is supported, including names that contain commas. |
.smiles, .smi, .txt | One SMILES value per nonblank row, or name<TAB>SMILES. Blank rows and rows beginning with # are ignored. |
Every parsed row counts as one ligand. A two-row CSV therefore requires Simultaneous co-docking or Batch docking; it cannot be submitted in Single ligand mode. Missing and malformed SMILES are reported against the source row.
Vina itself accepts an explicit search-box center and size. ProteinIQ provides four ways to define those native values:
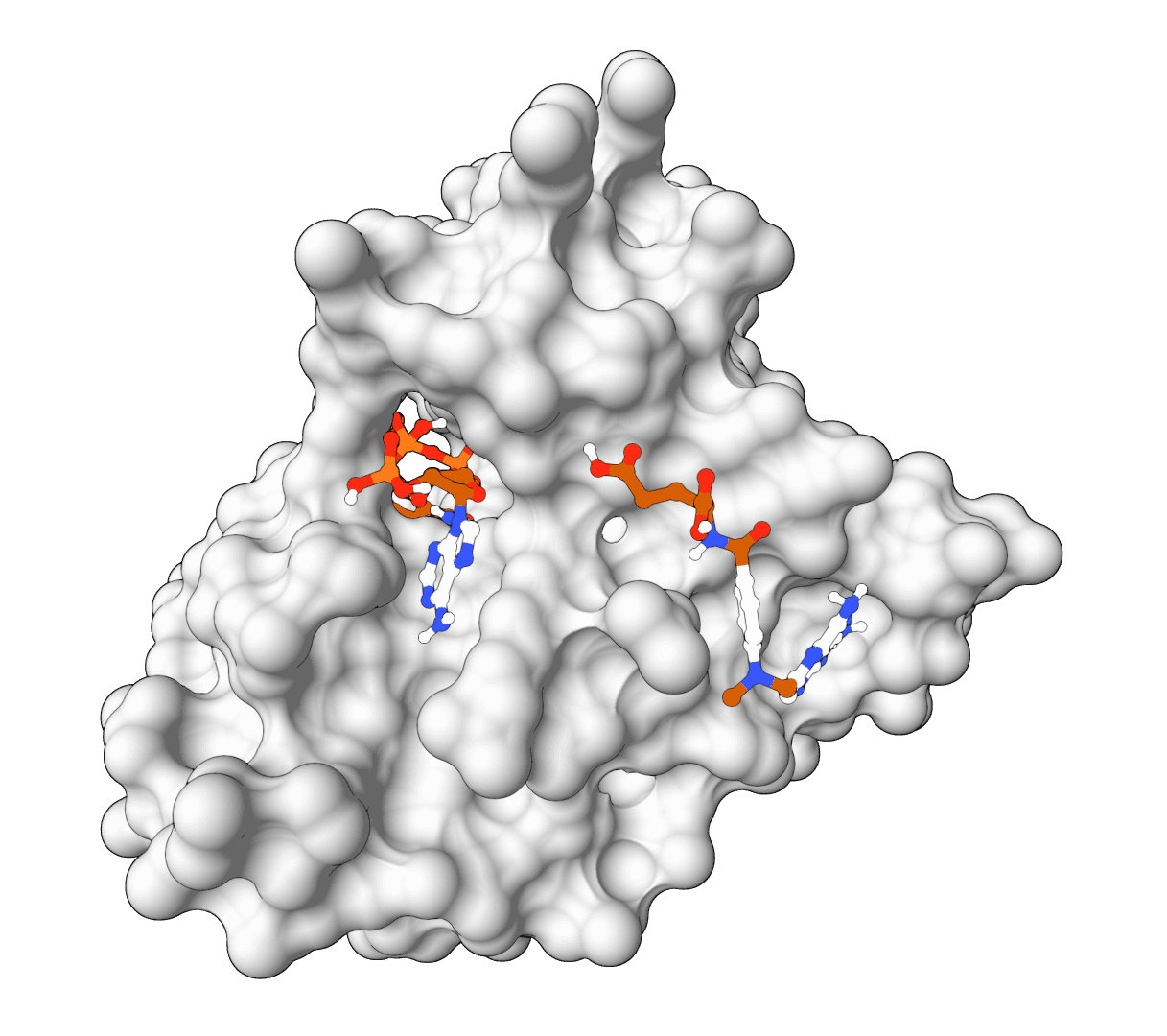
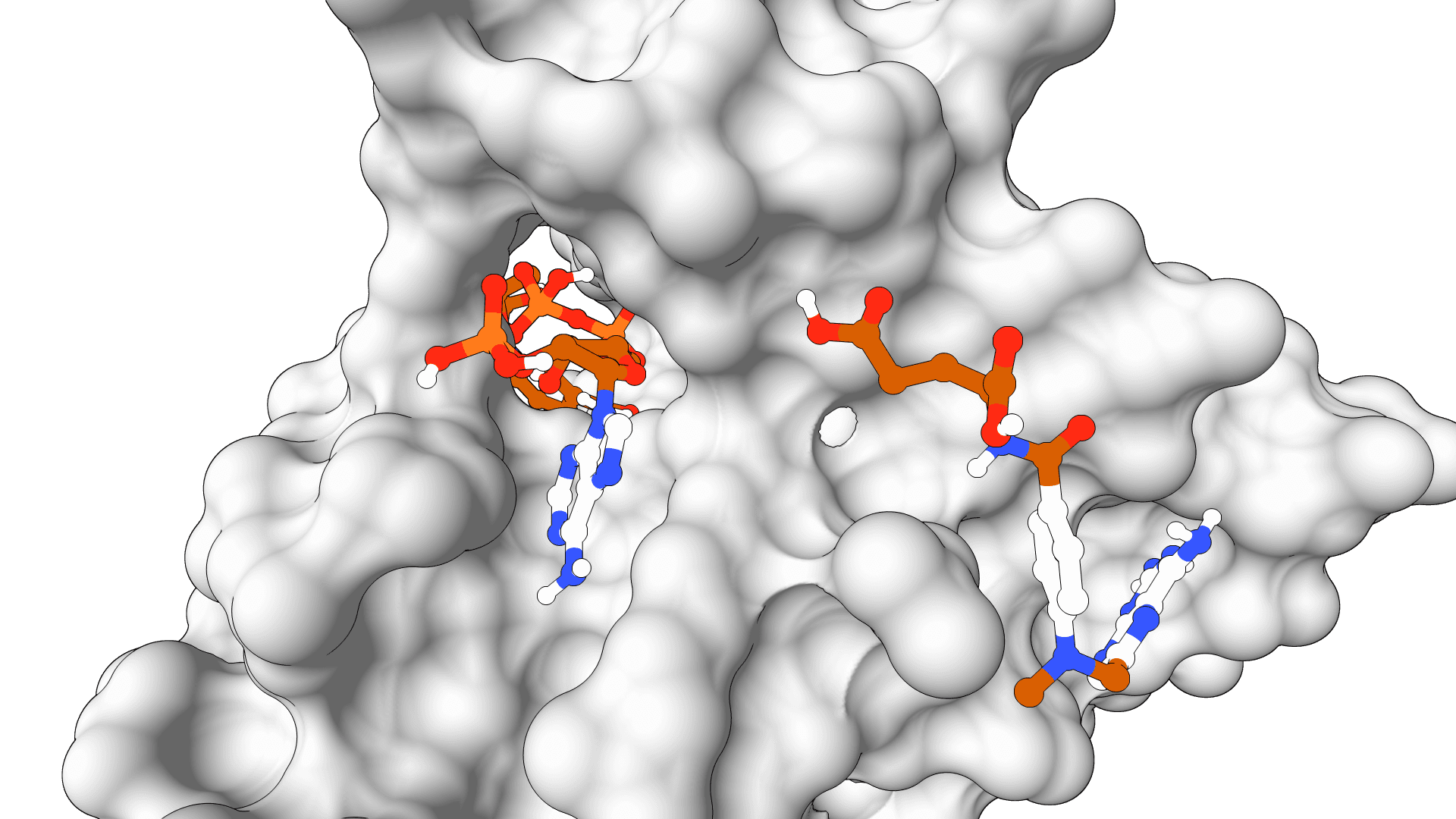
The interactive preview shows the receptor and computed box before submission. It supports drag rotation, mouse-wheel zoom, and atom selection to toggle residues in residue mode. The resolved method, source residue IDs, padding, center, size, volume, and enclosed atom count are saved in the result summary and run log.
For a receptor with a co-crystallized ligand, bound-ligand mode is the strongest starting point because it creates a focused box from experimental coordinates. A whole-protein box is less informative: enlarging the volume increases the conformational search burden, so the same Exhaustiveness value provides less sampling per region of space.
Automatic bound-ligand selection is available for source PDB, ENT, CIF, and mmCIF receptors. A prepared PDBQT is treated as an already finalized receptor, so selected residues, manual coordinates, or whole-protein bounds should be used with that input.
When no organic bound ligand is detected, the pocket method remains unselected. A residue selection, manual box, or whole-protein box must be chosen before submission.
The preparation route depends on the submitted receptor format:
| Receptor format | Preparation route |
|---|---|
| PDB or ENT | Guarded PDB2PQR preparation by default, including removal of crystallographic waters for a conventional water-free receptor, followed by Meeko receptor PDBQT generation. PDB2PQR can be disabled to run direct Meeko preparation. |
| CIF or mmCIF | Coordinate and connection normalization to PDB records, followed by the same guarded PDB2PQR and Meeko route. The original CIF/mmCIF remains available as the submitted source structure. |
| PDBQT | Used unchanged. PDB2PQR and Meeko are bypassed because the receptor is already prepared for docking. |
CIF/mmCIF normalization preserves compatible author atom, residue, and chain identifiers. Explicit covalent and metal connections are retained as PDB LINK records when they can be represented safely. PDB2PQR 3.7.1 then writes a native PDB intermediate containing repaired heavy atoms and its assigned hydrogens. ProteinIQ retains that complete intermediate for audit, removes its explicit hydrogen records before the next stage, and lets Meeko 0.7.1 assign the final hydrogens, Gasteiger charges, atom types, and receptor PDBQT. PDB2PQR force-field charges and radii are not passed to Vina.
Optional PROPKA protonation uses the selected receptor pH, with supported protonation states carried into Meeko through PDB2PQR's residue naming and heavy-atom geometry rather than its explicit hydrogen records. Standard preparation removes deposited waters with PDB2PQR and records every removed water residue; Retain crystallographic waters opts into using those waters as part of the fixed receptor model. Retained waters can affect docking and must be compatible with Meeko. This setting is distinct from hydrated ligand docking. Disabling Prepare with PDB2PQR sends the submitted or normalized PDB records directly to Meeko without this boundary transformation. ProteinIQ compares structures before and after preparation, records added hydrogens and missing heavy atoms, and stops rather than silently accepting unsupported structural changes.
Receptors with repeated chain, residue, and atom identities are blocked because merged or overlaid protein copies cannot be chosen safely. Trim structure can keep the intended coordinate segment; if both copies are required, the source structure needs unique chain IDs. PDBFixer can repair other structural problems before docking, but it cannot decide which overlaid copy should be retained.
The run also stops when PDB2PQR removes an existing heavy atom or residue, adds a residue, changes record types, performs an unexpected residue rename, or encounters multiple models, unresolved alternate conformers, or unexplained LINK or CONECT chemistry. When a receptor contains alternate conformers, set one Default alternate location or provide complete residue-specific choices such as A:42=B,B:17=A. ProteinIQ applies those explicit choices before pocket calculation and receptor preparation, and records every retained conformer and removed coordinate identity in the receptor preparation report. A parseable LINK may be removed when one of its referenced atoms is absent because it no longer describes chemistry in the submitted coordinates. Malformed links, unresolved CONECT records, modified residues, cofactors, metals, and other covalently linked components require an explicit preparation decision.
Bound-ligand pocket mode saves the selected ligand coordinates and removes that exact ligand before receptor preparation. The ligand-like cleanup, enabled by default, also recognizes two narrowly defined chain types:
ATOM and HETATM residues, has no more than 20 residues, and has an explicit LINK to a larger receptor chain.NH2 cap, has an explicit terminal C-N link, contains no other unexplained connectivity, and sits beside a larger receptor chain.The same cleanup removes orphaned LINK records when either referenced atom is absent, without changing coordinate records. It can remove the complete N3 inhibitor chain from 6LU7, remove endomorphin-1 from deposited 9WSW, and discard stale link metadata without treating ordinary short protein chains, metals, or standalone cofactors as ligands. Every automatic removal is recorded in receptor_preparation.json.
Every successful run returns receptor_preparation.json, the full receptor_preparation.log, input/output SHA-256 hashes, and the prepared receptor PDBQT. Runs with PDB2PQR enabled also return the complete native pdb2pqr_prepared_receptor.pdb and an inventory of added atoms, protonation-name changes, hydrogen counts, heavy-atom displacement, explicit hydrogen records removed before Meeko, and the SHA-256 hash of the actual Meeko input. Together, these files make preparation choices auditable and help keep receptor preparation consistent across a ligand series.
The default scoring values match Vina 1.2.7. Bound-ligand pocket detection is a ProteinIQ preparation convenience and is the default when a source PDB contains a detectable co-crystallized ligand.
The main form keeps the decisions needed for a normal docking run visible:
| Setting | Description |
|---|---|
Ligand mode | Runs one ligand on Free, up to 3 simultaneous or 5 batch ligands on Plus, and up to 5 simultaneous or 10 batch ligands on Pro. |
Scoring function | Uses Vina, Vinardo, or AutoDock4 scoring. AutoDock4 is required for hydrated and zinc-specific workflows. |
Exhaustiveness | Search thoroughness with a default of 8. Plan maxima are 8 on Free, 32 on Plus, and 64 on Pro. Higher values increase runtime and sampling. |
Number of poses | Maximum docked poses returned. Plan maxima are 9 on Free, 20 on Plus, and 50 on Pro. Every generated pose remains available in the results and files. |
Binding pocket | Defines the search from a bound ligand, selected residues, manual coordinates, or whole-protein bounds. Every mode resolves to Vina's native center and size values. |
One Advanced settings section contains the less common but still supported workflow decisions:
| Setting | Description |
|---|---|
Docking operation | Runs full docking, evaluates a placed pose with Score only, or refines a placed pose with Local only. |
Prepare with PDB2PQR | Enabled by default for PDB/ENT/CIF/mmCIF input. Runs guarded PDB2PQR preparation before Meeko. Prepared PDBQT input is always used unchanged. |
Use PROPKA protonation / Receptor pH | Optionally predicts standard-residue protonation states during PDB2PQR preparation at the selected pH. |
Retain crystallographic waters | Disabled by default. Enable only when deposited waters are intentionally part of the fixed receptor model. Every removed or retained-water policy is recorded. |
Remove detected ligand-like chains | Removes only a narrowly classified cross-linked ligand chain, terminally NH2-amidated peptide ligand, or orphaned LINK whose endpoint atom is absent, and records every removal. |
Flexible residues | Treats selected receptor side chains as flexible during docking. Plus supports up to 3 selected residues and Pro supports up to 10. |
Random seed | Uses a fixed integer for reproducible searches. 0 lets Vina select a seed. |
Hydrated ligand workflow | Runs Vina's hydrated AutoDock4 protocol for one ligand. |
Zinc metalloprotein mode | Runs the AutoDock4Zn pseudo-atom and zinc-parameter protocol. |
Specialized map controls, optimization limits, receptor-template overrides, destructive residue deletion, randomize-only execution, and custom scoring weights remain available for programmatic runs but are intentionally omitted from the standard form.
Upon successful docking, ProteinIQ presents the native Vina results in three formats:
| Output | Description |
|---|---|
Structure | Interactive 3D view of the receptor and docked pose geometry. |
Data | Dock mode returns affinity, RMSD lower and upper bounds, and all five native energy terms. Score and local modes return Vina's named eight-term energy vector. |
Files | Individual pose_modeNN.pdbqt structures, native combined PDBQT, prepared receptor and ligand PDBQT, optional PDB2PQR-prepared receptor PDB, receptor preparation report and complete preparation log, run summary, and hydrated dry or scored poses when applicable. Individual poses are also available as downstream workflow artifacts. |
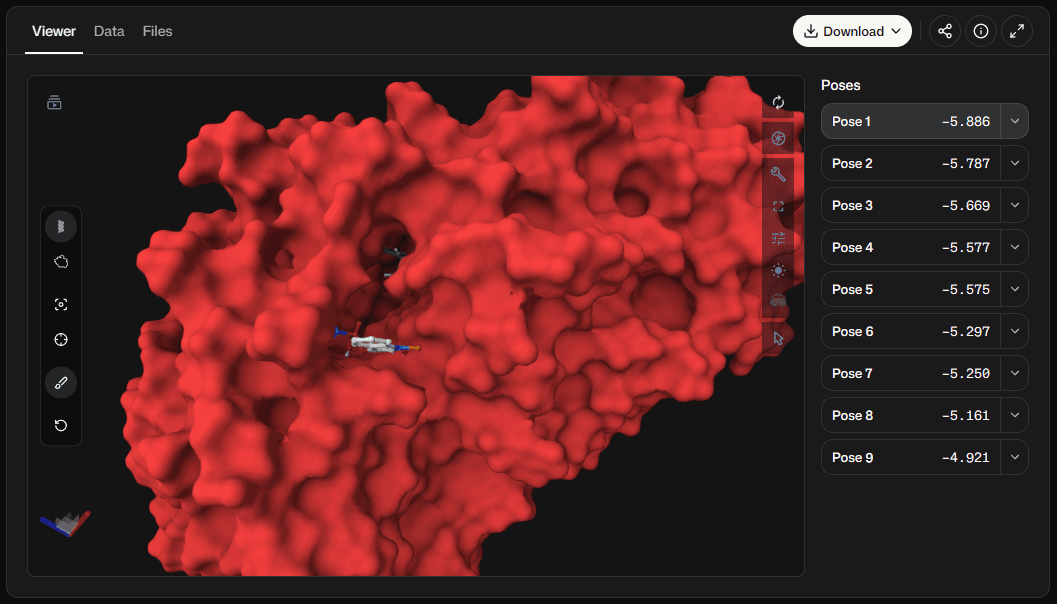
| Result column | How to read it |
|---|---|
Affinity (kcal/mol) | Vina's primary score for the pose. More negative values rank more favorably within the same scoring function and preparation protocol. |
RMSD lower bound (Å) | Symmetry-aware lower estimate of the pose's distance from the best-ranked returned mode. |
RMSD upper bound (Å) | Upper estimate of the same distance using the fixed atom correspondence. |
Intermolecular energy | Scored interaction between the movable ligand and the receptor. |
Intramolecular energy | Internal strain term for the movable atoms in the pose. |
Torsional energy | Ligand flexibility penalty used by the selected scoring function. |
Fifth native energy term | Vina or Vinardo reports the best-pose internal reference term; AutoDock4 reports the negative intramolecular term. The adjacent label identifies which meaning applies. |
The affinity score is most useful as a relative ranking within one consistent experiment. Absolute kcal/mol values should not be treated as direct binding free energies, especially across different targets, scoring functions, protonation states, or receptor preparations. Vina, Vinardo, and AutoDock4 scores should not be compared as though they share one scale.
The RMSD lower and upper bounds describe each returned mode's distance from the best-ranked returned mode. They do not measure accuracy against an experimental crystal pose unless that experimental reference has been aligned and evaluated separately. Similar RMSD values often identify a shared pose family, while a distinct cluster can represent an alternative binding hypothesis.
Pose geometry matters as much as score. A slightly worse score with sensible hydrogen bonding, steric fit, and ligand burial is often more credible than the top-ranked pose if that pose shows clashes or unrealistic exposure. For batch docking, comparisons are most meaningful when all ligands were prepared with the same protonation and tautomer assumptions.
Before screening new ligands against a receptor, a redocking control can test the setup. Remove a known co-crystallized ligand, dock that same ligand back into its experimental pocket, and compare the predicted pose with the crystal coordinates. Failure to recover a plausible pose under increased exhaustiveness points to the search box, receptor or ligand preparation, missing cofactors or waters, or a scoring limitation that should be resolved before interpreting a compound ranking.
ProteinIQ checks box/receptor overlap in the browser when coordinates are available and repeats the check against the prepared receptor before Vina runs. A box whose coordinate bounds do not overlap the prepared receptor is rejected. If the bounds overlap but contain no receptor atoms, the result includes a warning to verify that the box encloses the intended pocket.
Each pose_modeNN.pdbqt file can feed a compatible downstream workflow step. PoseBusters accepts SDF, MOL, MOL2, or PDB rather than PDBQT, so a selected Vina pose should first be converted to SDF with Open Babel, then passed to PoseBusters for geometry and chemical-validity checks.
This single-ligand example docks imatinib into the ATP-binding site of the c-Abl kinase domain, using the co-crystallized ligand in PDB 1IEP to define the search region.
1IEP (2.1 Å) and imatinib from PubChem CID 5291Random seed = 42 to make the stochastic search reproducible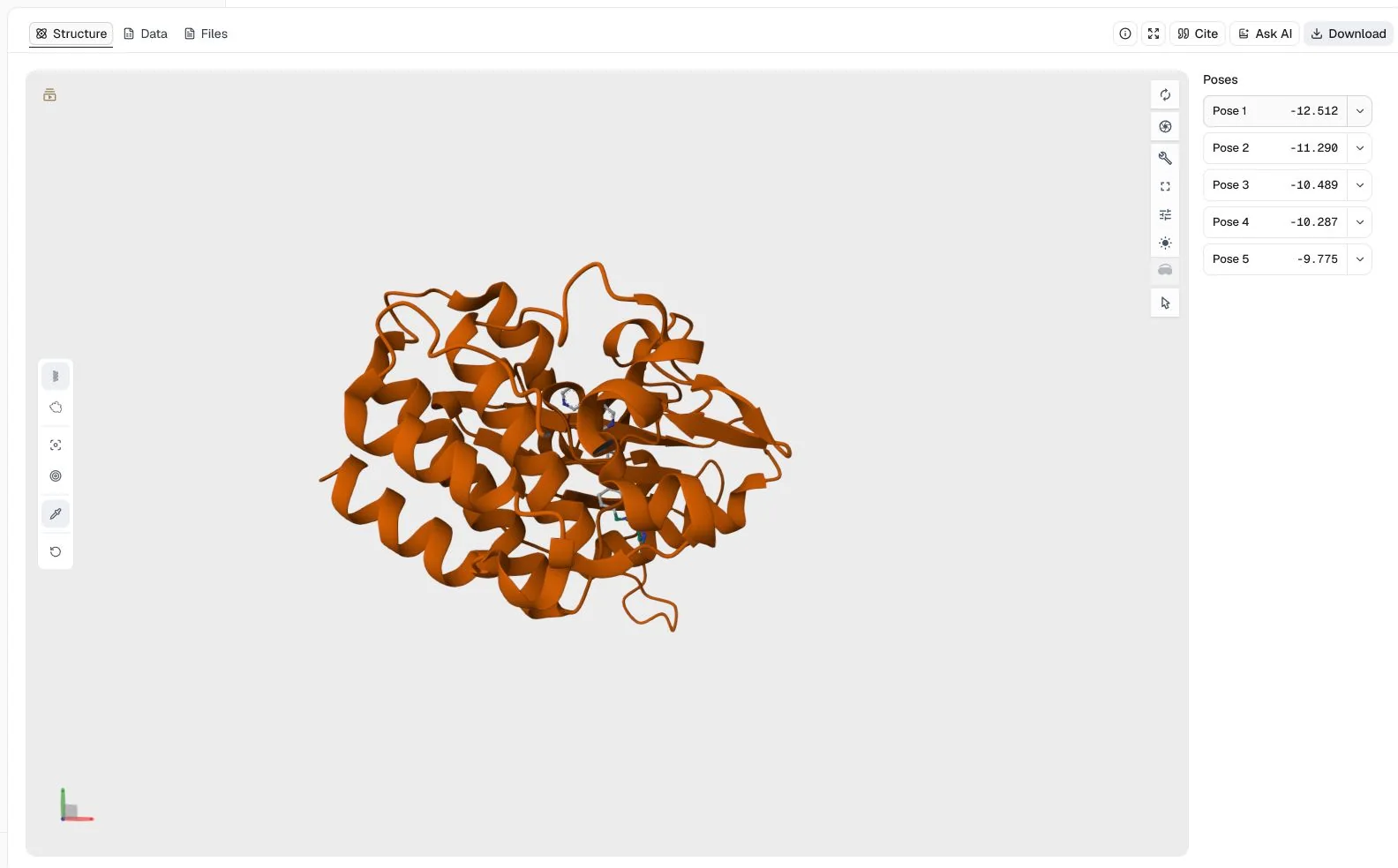
AutoDock Vina returned five poses in the configured energy window. The top-ranked pose has an Affinity of −12.512 kcal/mol, while the other visible poses range from −11.290 to −9.775 kcal/mol. The viewer supports inspection of the proposed placement in the kinase pocket, but the score is a docking ranking rather than an experimental binding free energy or proof that the pose is correct.
This example searches for joint placements of methotrexate and the NADPH cofactor in one human dihydrofolate reductase binding region. It demonstrates simultaneous co-docking, where both ligands move within the same Vina search rather than being docked as independent jobs.
1U72 (1.9 Å), methotrexate from PubChem CID 126941, and NADPH from PubChem CID 5884Ligand mode = Simultaneous co-docking; Binding pocket = Manual coordinates with center (26.624, 14.574, 5.689) Å and size (26.204, 28.087, 32.768) Å; Random seed = 42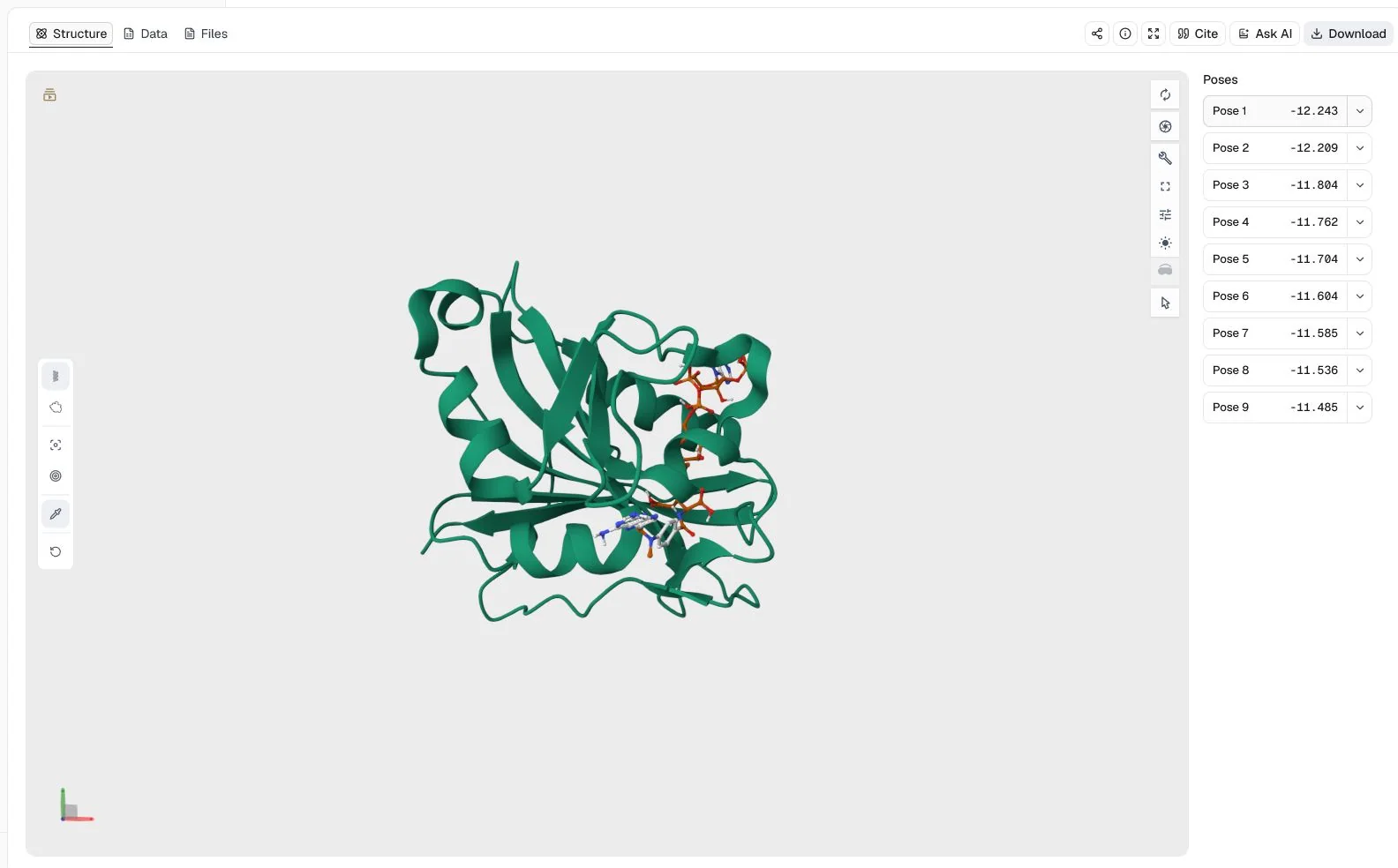
The structure view places both ligands in the DHFR receptor context and lists all nine joint poses. The top two poses score −12.243 and −12.209 kcal/mol, so their score difference is small even though their returned RMSD bounds indicate different joint geometries.
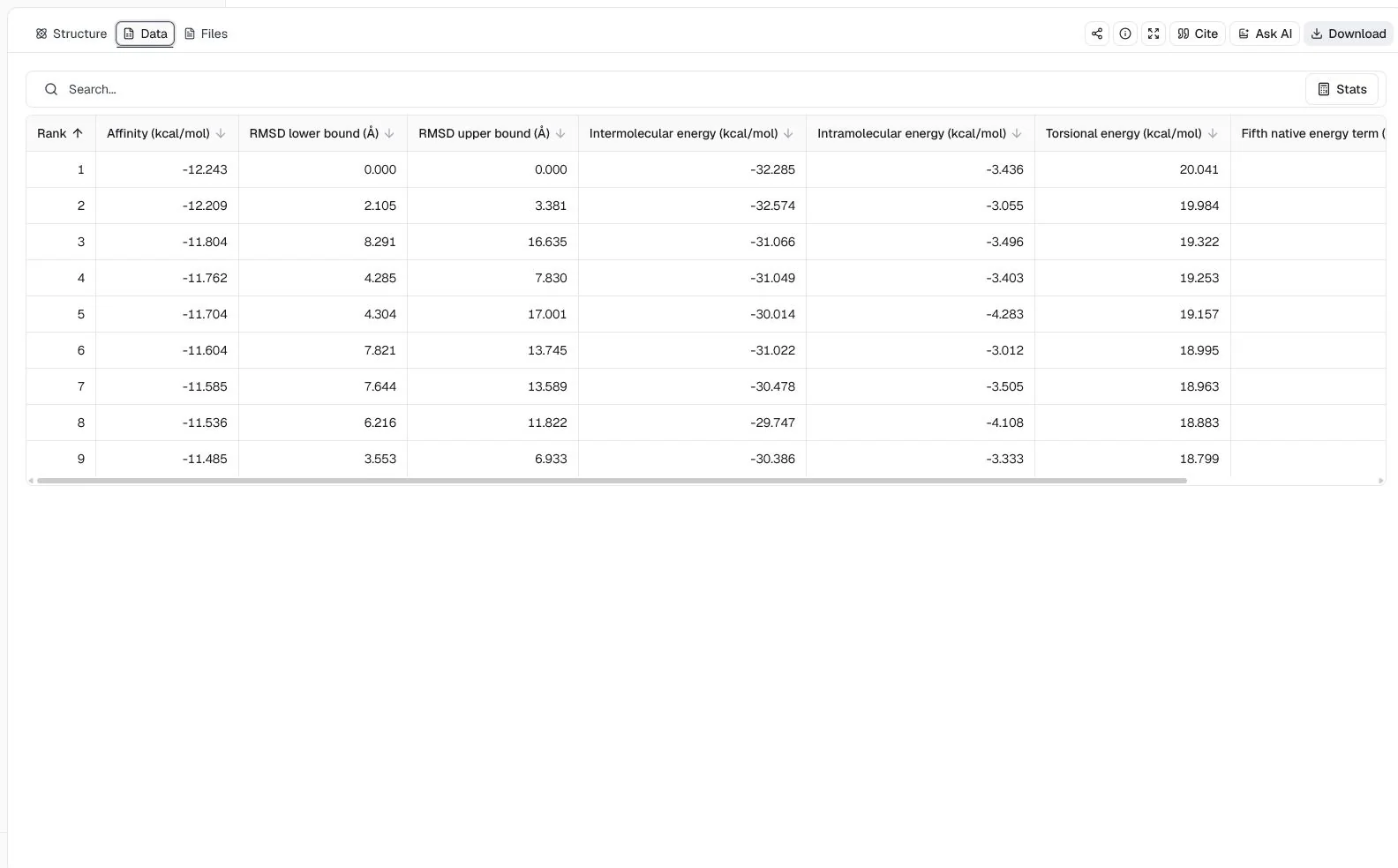
The Data view reports the best pose at −12.243 kcal/mol and the second pose with an RMSD lower bound of 2.105 Å and RMSD upper bound of 3.381 Å relative to the best returned mode. These values compare Vina's joint pose solutions; they do not establish that both molecules bind simultaneously in an experimental system.
This focused batch runs imatinib, nilotinib, and ponatinib independently against the same c-Abl receptor and search box. Holding the receptor preparation and Vina setup constant makes the returned scores useful for an initial within-run comparison.
1IEP (2.1 Å), with imatinib, nilotinib, and ponatinib as three independent ligandsLigand mode = Batch docking; Binding pocket = Manual coordinates with center (15.190, 53.902, 16.917) Å and size (18.664, 26.739, 23.526) Å; Random seed = 42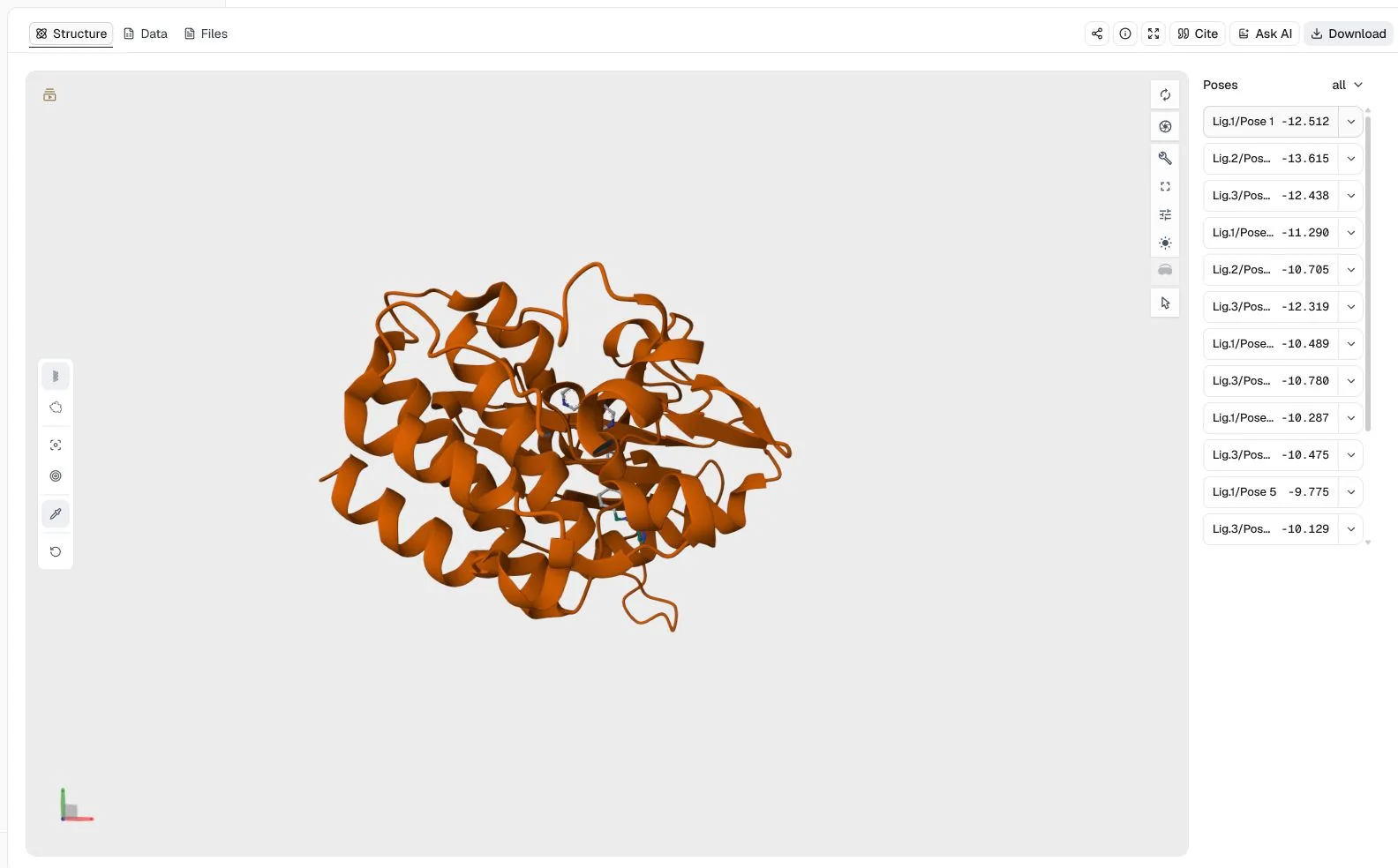
The structure view combines the 16 successful poses returned across the three ligands and lets each result be inspected in the shared c-Abl receptor context.
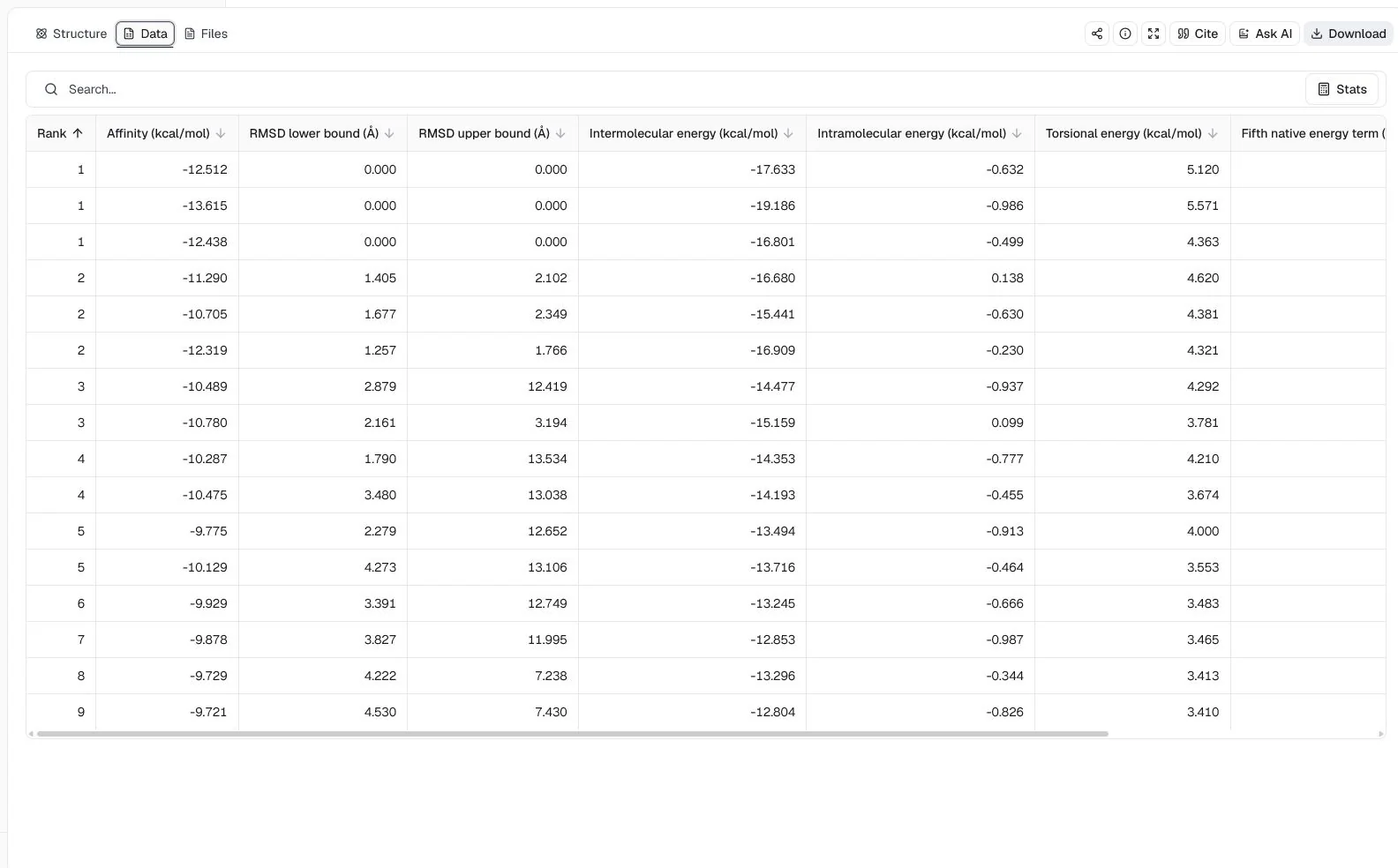
The leading Affinity values are −13.615 kcal/mol for nilotinib, −12.512 kcal/mol for imatinib, and −12.438 kcal/mol for ponatinib. Nilotinib ranks first under this specific preparation, search box, scoring function, and seed, but that ordering is a computational prioritization result, not a measured affinity series. It should be checked with pose inspection, repeated searches, and experimental evidence.
AutoDock Vina combines an empirical scoring function with stochastic global search and gradient-based local optimization. The ligand is translated, rotated, and flexed inside a predefined search volume; each candidate pose is scored; and promising conformations are refined before final clustering and ranking.
The scoring function estimates binding favorability from weighted steric, hydrophobic, hydrogen-bonding, and ligand-flexibility terms. Vina uses the default empirical model. Vinardo changes the atom-type potentials and weights. AutoDock4 uses map-based van der Waals, hydrogen-bonding, electrostatic, desolvation, and torsional terms, and is required for the zinc-specific and hydrated workflows.
Vina uses iterated local search, with candidate conformations initialized stochastically and refined by a BFGS local optimizer. Exhaustiveness controls the total independent search effort. Number of poses, Energy range, and the minimum RMSD separation determine which alternatives survive to the final report, so requesting more poses does not guarantee that Vina will return that many.
Most docking calculations keep the receptor rigid apart from ligand torsions. When Flexible residues are specified, selected receptor side chains are allowed to move during the search. This can recover poses that rigid docking would miss, but increases the search space and runtime substantially. Flexible docking is usually reserved for a few residues with a clear mechanistic rationale for moving.
| Goal | Suggested method |
|---|---|
| Dock into a known or user-defined pocket | AutoDock Vina for a faithful empirical docking search with explicit control over the box and preparation. |
| Add convolutional neural network rescoring | GNINA, which combines Vina-style sampling with CNN scoring and rescoring. |
| Propose poses without defining a search box | DiffDock for a learned, structure-wide pose proposal workflow. |
| Run larger AutoDock4 virtual screens on a GPU | AutoDock GPU for accelerated AutoDock4 searches. |
| Validate the chemical and geometric pose quality | Open Babel followed by PoseBusters after converting the selected PDBQT pose to a supported molecular file format. |