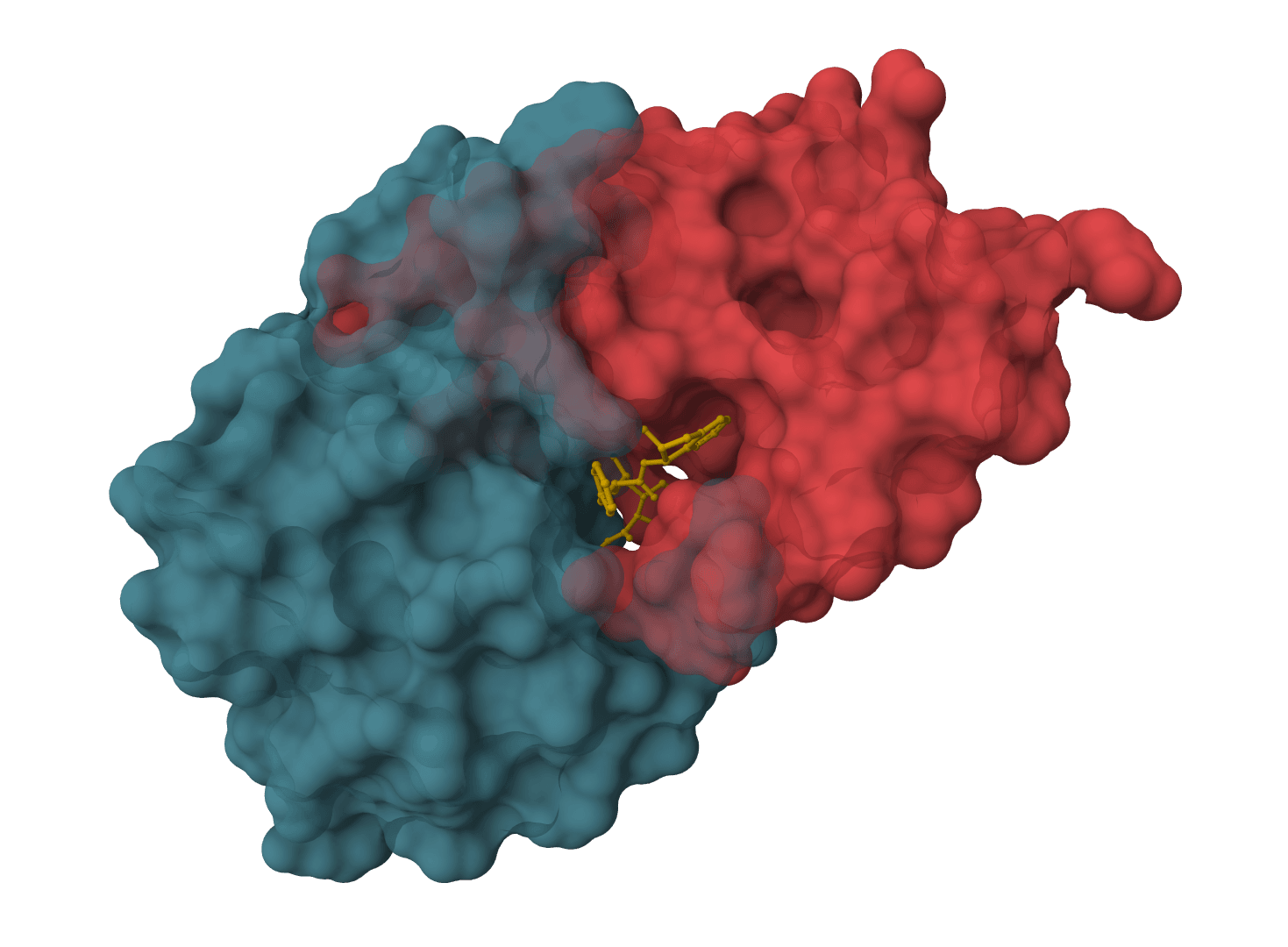
Enhanced molecular docking with custom scoring functions and fast minimization Learn more
Run
25 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job”.
Enhanced molecular docking with custom scoring functions and fast minimization Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
Enhanced molecular docking with custom scoring functions and fast minimization Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
SMINA is a fork of AutoDock Vina developed by David Koes at the University of Pittsburgh. It extends Vina with comprehensive support for custom scoring functions, dramatically faster minimization, and enhanced workflows for scoring function research.
Published in 2013, SMINA was designed to address limitations in Vina's scoring function development capabilities. The tool enables researchers to define custom scoring functions with user-specified weights, making it valuable for both standard docking and advanced scoring research.
SMINA also includes the Vinardo scoring function, an optimized variant that consistently outperforms Vina's default scoring in pose prediction, ranking, and virtual screening benchmarks.
SMINA inherits Vina's core search algorithm while extending its scoring capabilities. The search uses Iterated Local Search with BFGS quasi-Newton optimization, exploring the conformational space through random mutations of position, orientation, and torsion angles.
SMINA provides three scoring options:
Default (Vina) uses the original AutoDock Vina scoring function with Gaussian steric terms, hydrogen bonding, hydrophobic interactions, and a rotatable bond penalty. This is well-validated and suitable for most docking applications.
Vinardo removes Vina's problematic second Gaussian term that contributed 58% of binding energy despite creating artifacts. Vinardo uses optimized atomic radii and simplified terms, achieving 89.7% top-1 docking success versus Vina's 80.5% on the CASF-2013 benchmark.
Custom scoring allows researchers to define their own scoring functions using 58 available interaction terms, including electrostatics, desolvation, Lennard-Jones 4-8 potentials, and simple property counts.
SMINA's custom scoring supports these term types:
gauss), repulsion potentialsEach term accepts parameters for offset, width, cutoff, and exponent, enabling fine-grained control over the scoring function behavior.
SMINA provides four operational modes:
| Mode | Description | Speed |
|---|---|---|
Full docking | Global search + local optimization | Standard |
Minimize only | Refine existing pose without global search | 10-20x faster |
Score only | Evaluate pose without moving atoms | Fastest |
Local only | Local optimization from starting pose | Fast |
The Minimize only mode is particularly valuable for pose refinement workflows, achieving 10-20x speedup over full docking by skipping global search.
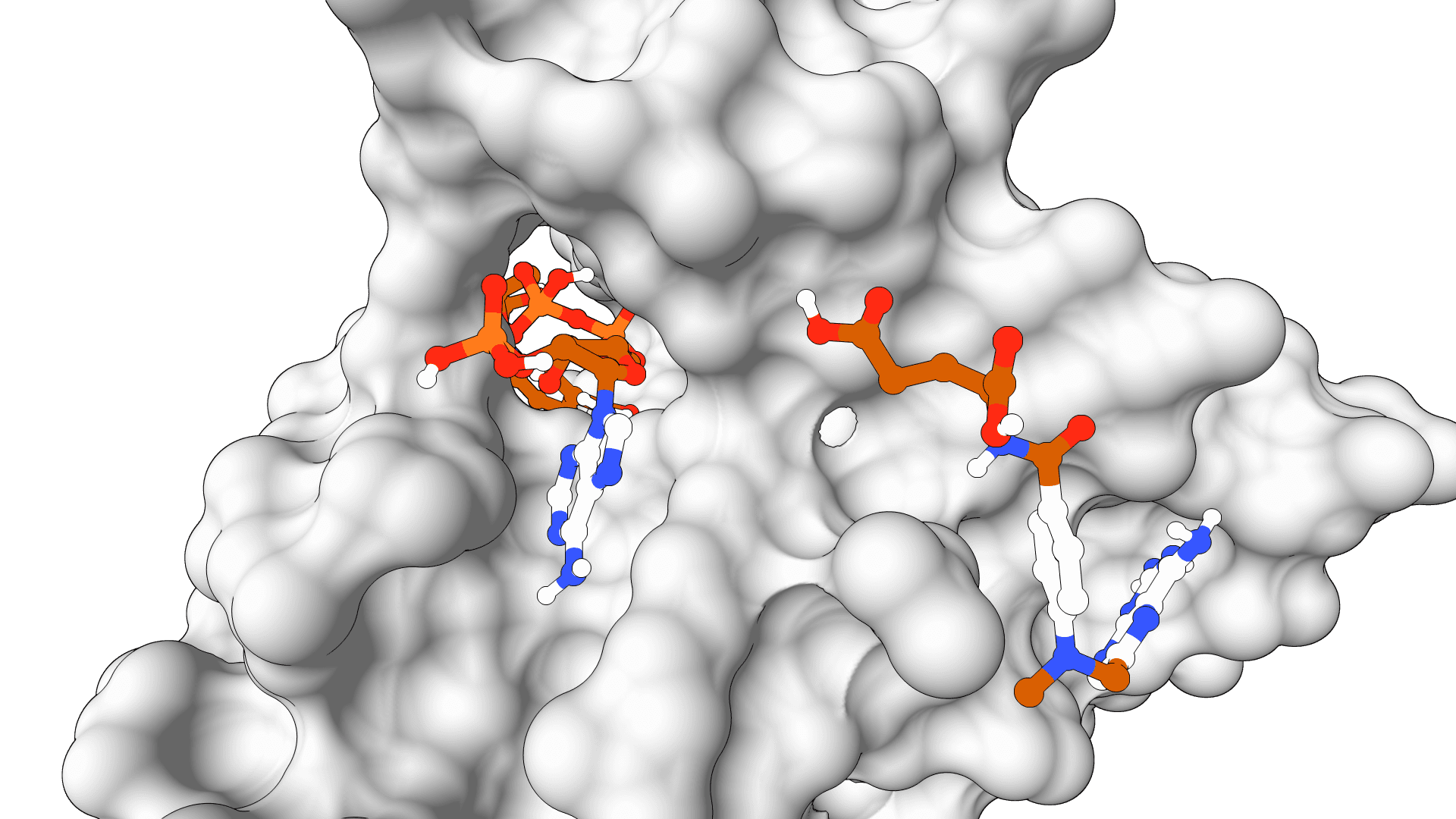
Upload a PDB file or enter an RCSB PDB ID. The protein should be prepared with hydrogens added and missing residues fixed. Use PDB Fixer for automated preparation.
Enter a SMILES string for small molecules, or upload an SDF, MOL, or PDBQT file. SMINA automatically generates 3D coordinates and calculates partial charges via OpenBabel.
For complex ligands (macrocycles, peptides, >150 atoms), pre-prepared PDBQT files are recommended to ensure proper handling.
The search box defines where SMINA explores ligand binding:
We recommend using autobox when a reference ligand structure is available, as this focuses the search and improves both speed and accuracy.
Affinity is reported in kcal/mol. More negative values indicate stronger predicted binding:
| Range | Interpretation |
|---|---|
| -4 to -6 | Weak binding |
| -6 to -8 | Moderate binding |
| -8 to -10 | Strong binding |
| < -10 | Very strong binding |
Vinardo scores may differ slightly from Vina due to the modified scoring function. Both use the same scale and interpretation guidelines.
SMINA ranks poses by predicted binding affinity. The top-ranked pose represents the most favorable binding mode, but examining multiple poses is recommended. Alternative binding modes within 1-2 kcal/mol of the best score may be biologically relevant.
On the CASF-2013 benchmark, Vinardo achieves:
r = 0.601 between predicted and experimental binding affinities| Feature | SMINA | AutoDock Vina | GNINA |
|---|---|---|---|
| Scoring | Vina/Vinardo/Custom | Vina | Vina + CNN |
| Max poses | 50 | 20 | 20 |
| Minimization speed | 10-20x faster | Standard | Standard |
| Custom scoring | Yes | No | No |
| Per-atom energies | Yes | No | No |
SMINA is ideal when you need custom scoring functions, fast minimization, or more than 20 poses. For standard docking with known scoring, Vina or GNINA work equally well. For protein-ligand docking with affinity-oriented pose ranking, compare PandaDock.
Custom scoring functions are defined as weighted sums of interaction terms. Each line specifies a weight and term with parameters:
-0.035579 gauss(o=0,_w=0.5,_c=8)
-0.005156 gauss(o=3,_w=2,_c=8)
0.840245 repulsion(o=0,_c=8)
-0.035069 hydrophobic(g=0.5,_b=1.5,_c=8)
-0.587439 non_dir_h_bond(g=-0.7,_b=0,_c=8)
1.923 num_tors_divParameters include:
o: Offset from optimal distancew: Gaussian widthc: Distance cutoffg: Good distance thresholdb: Bad distance thresholdDetailed term documentation is available in the SMINA GitHub repository.
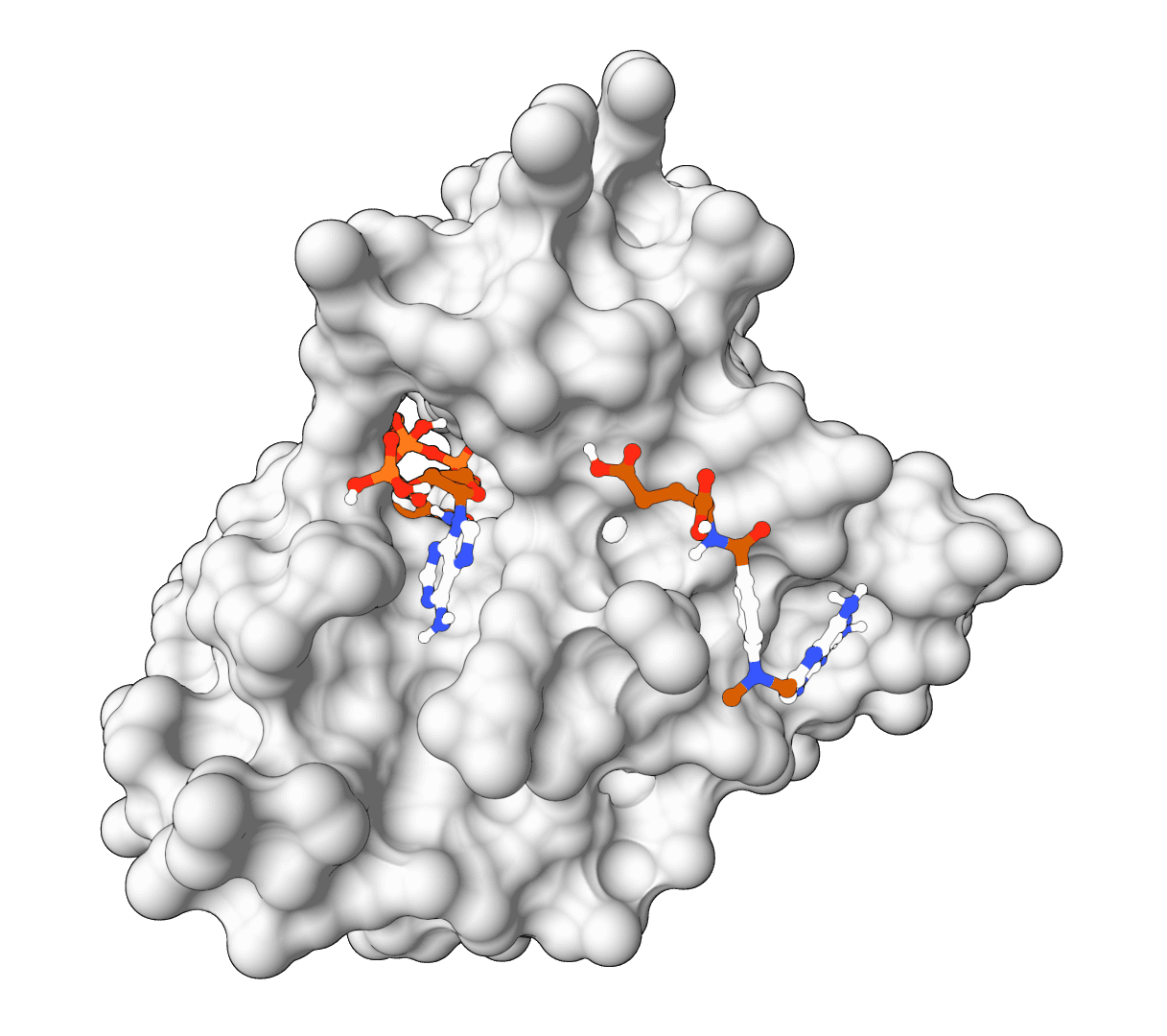
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
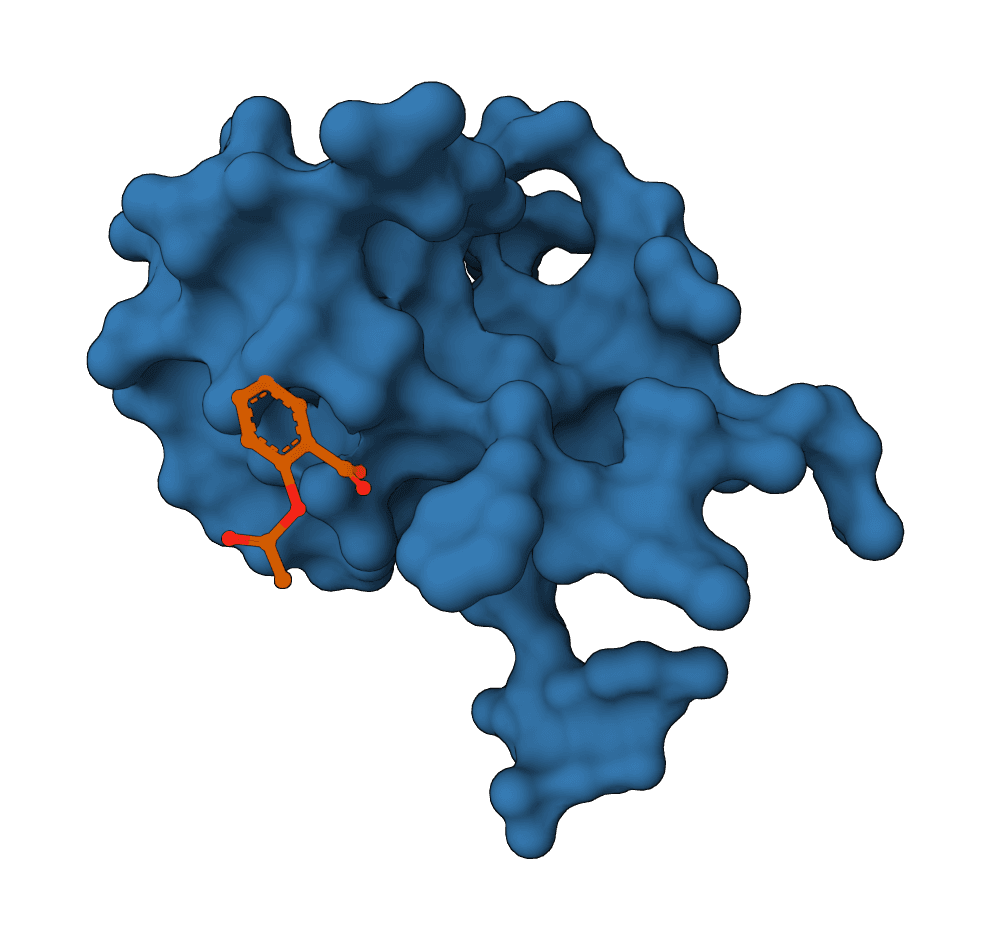
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
SMINA is a fork of AutoDock Vina developed by David Koes at the University of Pittsburgh. It extends Vina with comprehensive support for custom scoring functions, dramatically faster minimization, and enhanced workflows for scoring function research.
Published in 2013, SMINA was designed to address limitations in Vina's scoring function development capabilities. The tool enables researchers to define custom scoring functions with user-specified weights, making it valuable for both standard docking and advanced scoring research.
SMINA also includes the Vinardo scoring function, an optimized variant that consistently outperforms Vina's default scoring in pose prediction, ranking, and virtual screening benchmarks.
SMINA inherits Vina's core search algorithm while extending its scoring capabilities. The search uses Iterated Local Search with BFGS quasi-Newton optimization, exploring the conformational space through random mutations of position, orientation, and torsion angles.
SMINA provides three scoring options:
Default (Vina) uses the original AutoDock Vina scoring function with Gaussian steric terms, hydrogen bonding, hydrophobic interactions, and a rotatable bond penalty. This is well-validated and suitable for most docking applications.
Vinardo removes Vina's problematic second Gaussian term that contributed 58% of binding energy despite creating artifacts. Vinardo uses optimized atomic radii and simplified terms, achieving 89.7% top-1 docking success versus Vina's 80.5% on the CASF-2013 benchmark.
Custom scoring allows researchers to define their own scoring functions using 58 available interaction terms, including electrostatics, desolvation, Lennard-Jones 4-8 potentials, and simple property counts.
SMINA's custom scoring supports these term types:
gauss), repulsion potentialsEach term accepts parameters for offset, width, cutoff, and exponent, enabling fine-grained control over the scoring function behavior.
SMINA provides four operational modes:
| Mode | Description | Speed |
|---|---|---|
Full docking | Global search + local optimization | Standard |
Minimize only | Refine existing pose without global search | 10-20x faster |
Score only | Evaluate pose without moving atoms | Fastest |
Local only | Local optimization from starting pose | Fast |
The Minimize only mode is particularly valuable for pose refinement workflows, achieving 10-20x speedup over full docking by skipping global search.
Upload a PDB file or enter an RCSB PDB ID. The protein should be prepared with hydrogens added and missing residues fixed. Use PDB Fixer for automated preparation.
Enter a SMILES string for small molecules, or upload an SDF, MOL, or PDBQT file. SMINA automatically generates 3D coordinates and calculates partial charges via OpenBabel.
For complex ligands (macrocycles, peptides, >150 atoms), pre-prepared PDBQT files are recommended to ensure proper handling.
The search box defines where SMINA explores ligand binding:
We recommend using autobox when a reference ligand structure is available, as this focuses the search and improves both speed and accuracy.
Affinity is reported in kcal/mol. More negative values indicate stronger predicted binding:
| Range | Interpretation |
|---|---|
| -4 to -6 | Weak binding |
| -6 to -8 | Moderate binding |
| -8 to -10 | Strong binding |
| < -10 | Very strong binding |
Vinardo scores may differ slightly from Vina due to the modified scoring function. Both use the same scale and interpretation guidelines.
SMINA ranks poses by predicted binding affinity. The top-ranked pose represents the most favorable binding mode, but examining multiple poses is recommended. Alternative binding modes within 1-2 kcal/mol of the best score may be biologically relevant.
On the CASF-2013 benchmark, Vinardo achieves:
r = 0.601 between predicted and experimental binding affinities| Feature | SMINA | AutoDock Vina | GNINA |
|---|---|---|---|
| Scoring | Vina/Vinardo/Custom | Vina | Vina + CNN |
| Max poses | 50 | 20 | 20 |
| Minimization speed | 10-20x faster | Standard | Standard |
| Custom scoring | Yes | No | No |
| Per-atom energies | Yes | No | No |
SMINA is ideal when you need custom scoring functions, fast minimization, or more than 20 poses. For standard docking with known scoring, Vina or GNINA work equally well. For protein-ligand docking with affinity-oriented pose ranking, compare PandaDock.
Custom scoring functions are defined as weighted sums of interaction terms. Each line specifies a weight and term with parameters:
-0.035579 gauss(o=0,_w=0.5,_c=8)
-0.005156 gauss(o=3,_w=2,_c=8)
0.840245 repulsion(o=0,_c=8)
-0.035069 hydrophobic(g=0.5,_b=1.5,_c=8)
-0.587439 non_dir_h_bond(g=-0.7,_b=0,_c=8)
1.923 num_tors_divParameters include:
o: Offset from optimal distancew: Gaussian widthc: Distance cutoffg: Good distance thresholdb: Bad distance thresholdDetailed term documentation is available in the SMINA GitHub repository.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.