Analysis
AbLang
Restore missing residues in antibody sequences using a language model trained on the Observed Antibody Space (OAS) database. Achieves better restoration than IMGT germlines or ESM-1b while being 7x faster.
AbLang-2
Antibody-specific language model for predicting non-germline residues (NGL) in antibody sequences. AbLang-2 addresses germline bias in existing antibody language models by focusing on somatic hypermutation patterns, enabling more accurate prediction of amino acid likelihoods and generation of context-aware embeddings for antibody sequences.
AF-Cluster
Cluster Multiple Sequence Alignments to predict alternative protein conformations with AlphaFold2. Uses DBSCAN clustering to identify sequence subgroups.
Aggrescan3D
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
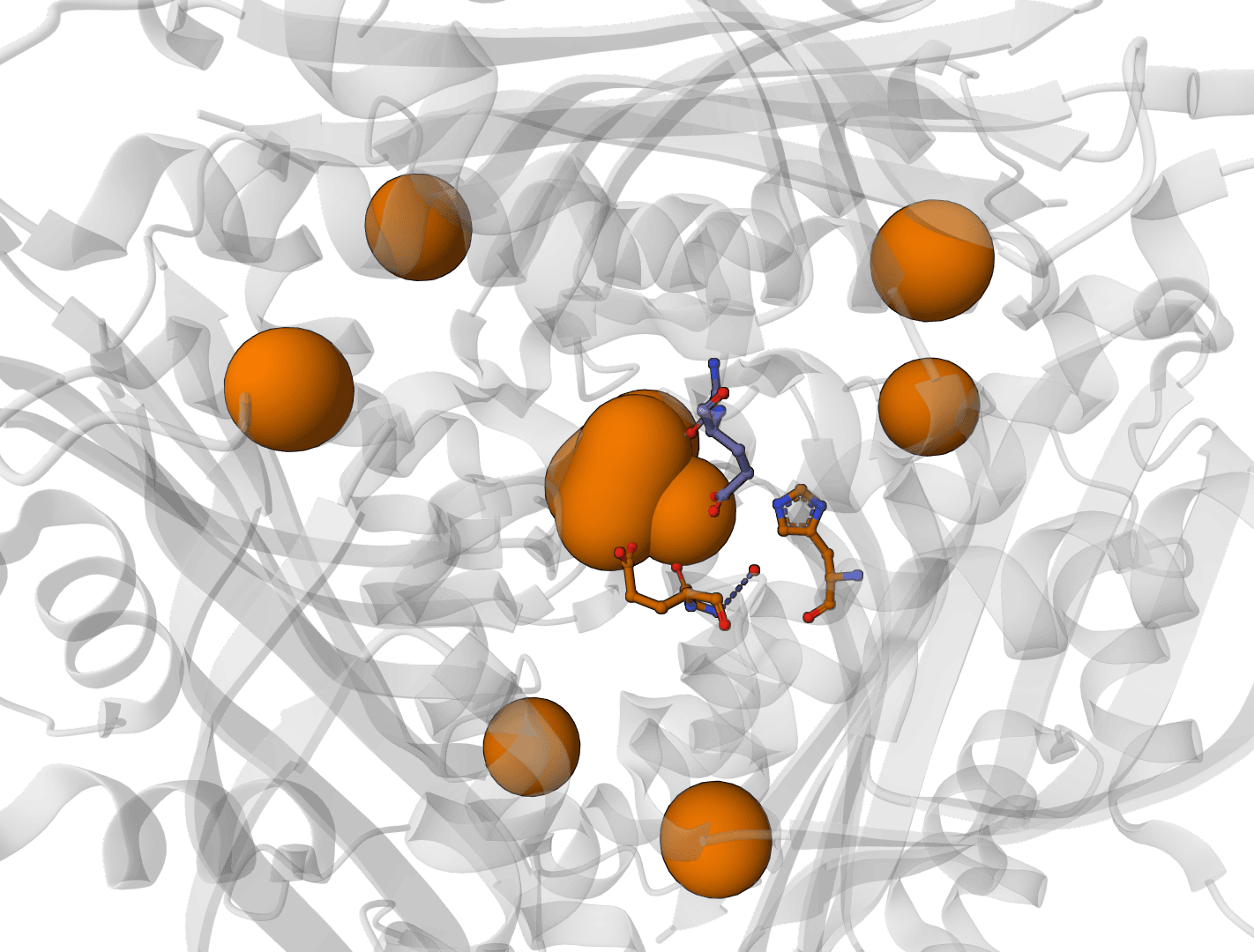
AllMetal3D
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).

AlphaGenome
AlphaGenome predicts variant effects on gene expression by comparing reference and alternate alleles. ProteinIQ currently supports RNA-seq analysis windows up to 512K base pairs. Uses your own DeepMind API key - no credit cost.
CANYA
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
Carbon
Carbon is a DNA language model for generation, scoring, and sequence comparison using the native Hugging Face Carbon model family.
Chou-Fasman
Predict protein secondary structure using the classic Chou-Fasman algorithm based on amino acid propensities
CleaveNet
Official CleaveNet tool for matrix metalloproteinase cleavage prediction and peptide generation. Predict cleavage z-scores plus uncertainty across 18 MMP variants, evaluate against truth z-scores, or generate candidate peptides unconditionally or from MMP z-score profiles.
CpG Island Finder
Identify CpG islands in DNA sequences using the Gardiner-Garden and Frommer criteria. Analyze GC content, CpG density, and observed/expected ratios.
DeepEMhancer
DeepEMhancer is a deep learning-based post-processing tool for cryo-EM maps. It performs automatic sharpening, masking, and denoising in a single step without requiring an atomic model. Supports half-map inputs for improved local mask estimation.
DeepImmuno
Predict peptide immunogenicity with DeepImmuno-CNN from peptide sequences and HLA alleles.
DockQ
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
DR-BERT
DR-BERT is a compact protein language model that predicts intrinsically disordered regions (IDRs) in proteins. It outputs per-residue disorder probability scores (0–1) from amino acid sequences, enabling fast and accurate annotation of disordered regions without structural data.
DSSP
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.
ESM-2
ESM-2 is a 650M parameter protein language model from Meta AI trained on 250M protein sequences. Generate rich sequence representations for downstream tasks like structure prediction, function annotation, and variant effect prediction.
ESM-C
ESM-C generates protein sequence representations and optional masked-token logits using Biohub protein language models. It supports the 300M, 600M, and 6B model variants for embedding extraction from canonical amino acid sequences.
FindPept
Match experimental peptide masses against theoretical digest fragments of a protein sequence. Identify peptides from mass spectrometry data by peptide mass fingerprinting.
GC content calculator
Calculate GC content, GC/AT skew, melting temperature, and CpG islands for DNA/RNA sequences, with a sliding-window GC plot. Analyze individual sequences or get combined statistics.
Hydropathy plot
Generate Kyte-Doolittle hydropathy plots to visualize hydrophobic and hydrophilic regions along protein sequences. Identify transmembrane domains and surface-exposed regions.
Hydrophobicity plot
Generate hydrophobicity plots using 24 different amino acid scales. Visualize hydrophobic and hydrophilic regions for protein analysis, epitope prediction, and membrane protein studies.
IPC 2.0 (isoelectric point calculator)
Isoelectric Point Calculator 2.0 - Predict protein/peptide isoelectric point (pI) using 18+ validated pKa scales, SVR models, and deep learning. Supports proteins, peptides, and comprehensive analysis.
IPSAE
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
LocScale
LocScale performs physics-informed local sharpening of cryo-EM density maps using half-maps or full MRC/MAP volumes, with optional mask and reference-map inputs.
Molecular descriptors
Compute 200+ RDKit molecular descriptors, drug-likeness rule violations, and structural fingerprints for QSAR, virtual screening, and ML workflows
MolProbity
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
ORF Finder
Find all Open Reading Frames (ORFs) in DNA sequences. Searches all six reading frames and supports multiple genetic codes.
PDBsum
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Peptide cutter
Predict protease and chemical cleavage sites across a protein sequence for up to 39 enzymes simultaneously. Identify where each enzyme cuts, the cleavage residue, and context window around each site.
Peptide mass calculator
Cleave a protein sequence with a chosen protease and compute the masses of the resulting peptides. Supports multiple enzymes, missed cleavages, chemical modifications, and different ion types for mass spectrometry experiment planning.
PLIP
Analyze noncovalent interactions in protein-ligand complex structures with PLIP, including hydrogen bonds, hydrophobic contacts, pi interactions, salt bridges, water bridges, halogen bonds, and metal complexes.
PoseBusters
PoseBusters validates generated or docked molecular poses with chemically and structurally grounded quality checks for molecular geometry, intermolecular interactions, and optional reference-pose agreement.
PPAP
PPAP (Protein-Protein Affinity Predictor) predicts binding affinity (ΔG and Kd) between interacting protein chains using deep learning with ESM2-3B embeddings. Requires a PDB with 2+ protein chains. Note: This tool is for protein-protein interactions only, not protein-ligand binding.
ProLIF
ProLIF calculates protein-ligand interaction fingerprints from 3D structures, returning residue-level interaction tables, interaction metadata, and native fingerprint files.
PROPKA 3
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
ProstT5
ProstT5 is a protein language model that bidirectionally translates between amino acid sequences and 3Di structural tokens. It enables fast structure-based searches and inverse folding by encoding structural information into a sequence-like representation.
Protein charge plot
Plot net charge vs pH for protein sequences. Visualize how protein charge changes across pH 0-14 and identify the isoelectric point (pI) where the net charge crosses zero.
Protein parameters
Calculate protein parameters, including molecular weight, theoretical pI, extinction coefficients, aromaticity, secondary structure fractions, atomic composition, estimated half-life, and several indices, including instability, aliphatic index, and GRAVY.
Protein scale profiler
Generate amino acid property profiles using 42 different scales spanning hydrophobicity, secondary structure propensity, flexibility, polarity, surface accessibility, antigenicity, and more.
Protein stability
Predict protein stability using validated BioPython methods: Instability Index, Aliphatic Index, GRAVY, flexibility analysis, and charge distribution
Protein-Sol
Predict protein solubility from amino acid sequence using the University of Manchester Protein-Sol method.
pySCA
Statistical Coupling Analysis for protein families. Identifies co-evolving residue groups (sectors) from multiple sequence alignments using the SCA method from the Ranganathan Lab.
Radius of gyration
Calculate the radius of gyration (Rg) for protein structures from PDB files. Supports multiple chains and atom selection options.
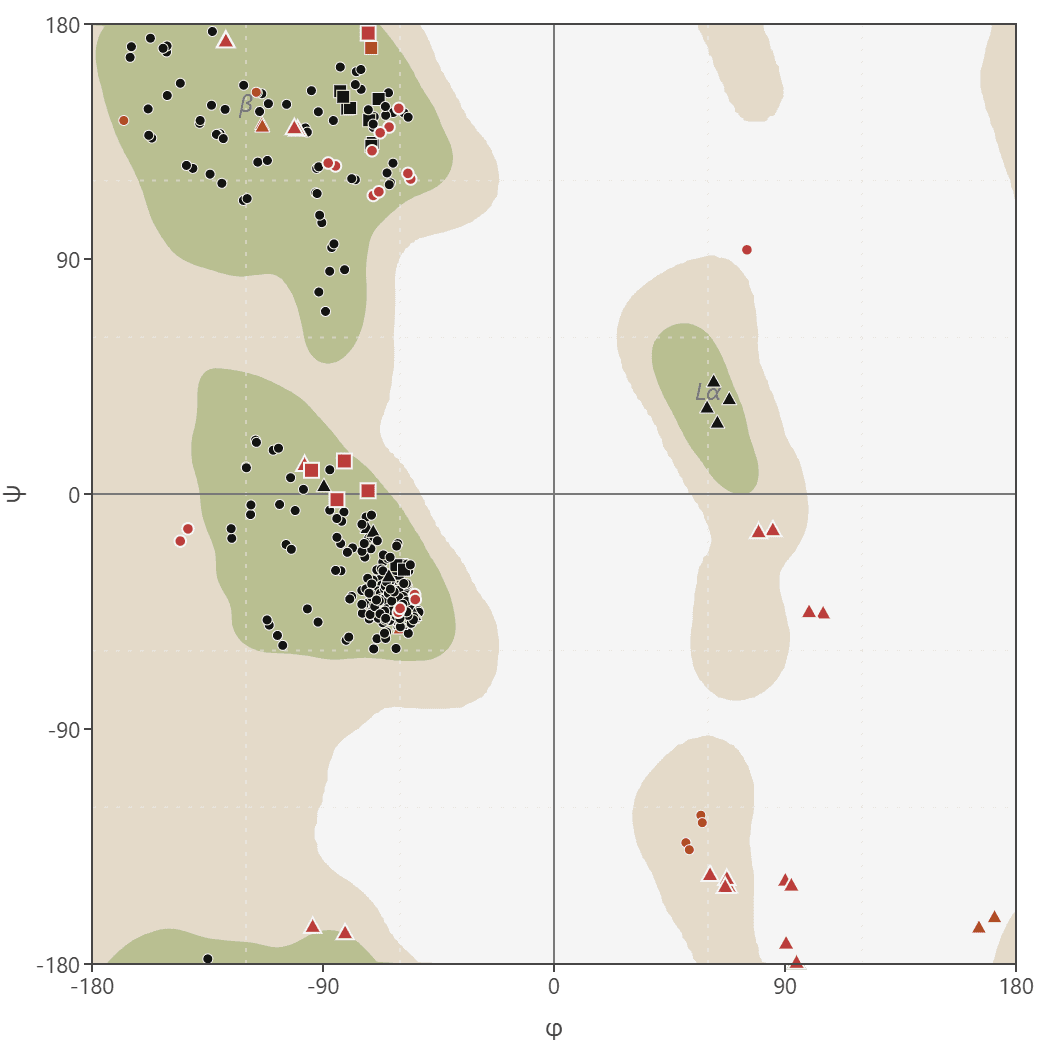
Ramachandran plot
Generate Ramachandran plots from PDB structures to analyze protein backbone dihedral angles (phi/psi). Visualize favored, allowed, and outlier regions.
RAxML-NG
Perform maximum-likelihood phylogenetic tree inference with RAxML-NG for aligned protein or DNA sequences. Supports ML search, bootstrap analysis, and native automatic model-family selection.
RMSD calculator
Calculate Root Mean Square Deviation (RMSD) between protein structures. Compare a reference PDB against multiple structures with automatic Kabsch alignment.
RNAcofold
RNAcofold predicts the joint secondary structure of two interacting RNA molecules and optionally reports partition-function and concentration-dependent equilibrium metrics.
RNAdistance
RNAdistance compares RNA secondary structures using the selected native ViennaRNA distance representation and comparison mode.
RNAdos
RNAdos calculates density-of-states summaries for RNA sequences, reporting representative structures and state counts across energy bands.
RNAduplex
RNAduplex computes the hybridization structure between two RNA sequences. Predicts the optimal duplex formation and binding energy.
RNAeval
RNAeval calculates the free energy of an RNA secondary structure for a given sequence. Evaluates if a proposed structure is thermodynamically favorable.
RNAfold
RNAfold predicts RNA secondary structure using minimum free energy (MFE) algorithms and optionally returns partition-function ensemble metrics when explicitly enabled.
RNALfold
RNALfold reports locally stable RNA secondary structures within a sliding window and returns their start and end positions on the input sequence.
RNAplex
RNAplex predicts fast query-target RNA interactions, reporting parsed hit coordinates, structures, and energies.
RNAplfold
RNAplfold computes local base pair probabilities using a sliding window approach. Useful for analyzing accessibility and identifying binding sites in long RNA sequences.
RNAplot
RNAplot renders ViennaRNA secondary-structure plot files from a supplied RNA sequence and dot-bracket structure.
RNAsubopt
RNAsubopt enumerates all RNA secondary structures within a specified energy range above the minimum free energy (MFE). Useful for exploring the structural ensemble and identifying alternative conformations.
RNAup
RNAup predicts accessibility-aware RNA-RNA interactions, reporting opening-energy terms alongside interaction energies and downloadable native output files.
SASA calculator
Calculate Solvent Accessible Surface Area (SASA) for protein structures using the Shrake-Rupley algorithm.
ScanNet
Geometric deep learning model for predicting protein binding sites directly from 3D structure. Identifies where proteins interact with other proteins, antibodies, or disordered proteins with high accuracy, including for novel protein folds.
SuperWater
Predict protein hydration sites from a structure using a diffusion model with ESM features and a confidence-filtering head.
ThermoMPNN
Predict protein thermostability changes (ΔΔG) for point mutations using a graph neural network. Enables computational saturation mutagenesis screening to identify stabilizing mutations.
TLimmuno2
Predict MHC class II peptide immunogenicity (CD4+ T cell response) using transfer learning with LSTM.
ViennaRNA
ViennaRNA supports a curated set of scientifically faithful ViennaRNA 2.7.2 workflows for RNA folding, density-of-states analysis, interaction prediction, local accessibility, plotting, inverse folding, and structure analysis.