
Predict protein stability from instability index, hydrophobicity, charge, and secondary-structure propensities. Learn more
Predict protein stability from instability index, hydrophobicity, charge, and secondary-structure propensities. Learn more
Predict protein stability from instability index, hydrophobicity, charge, and secondary-structure propensities. Learn more
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.
Predict protein thermostability changes (ΔΔG) for point mutations using a graph neural network. Enables computational saturation mutagenesis screening to identify stabilizing mutations.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
Predict protein solubility from amino acid sequence using the University of Manchester Protein-Sol method.
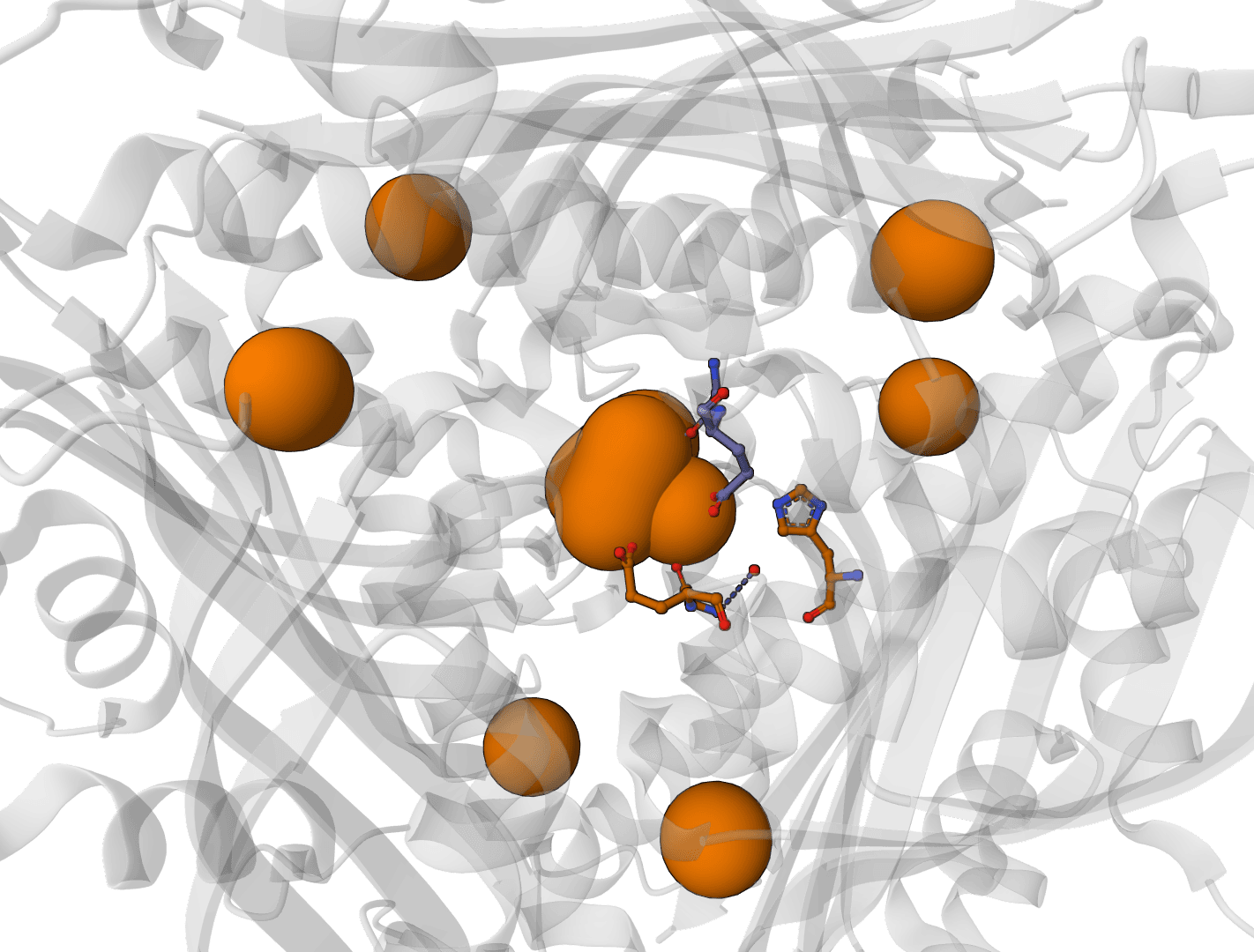
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
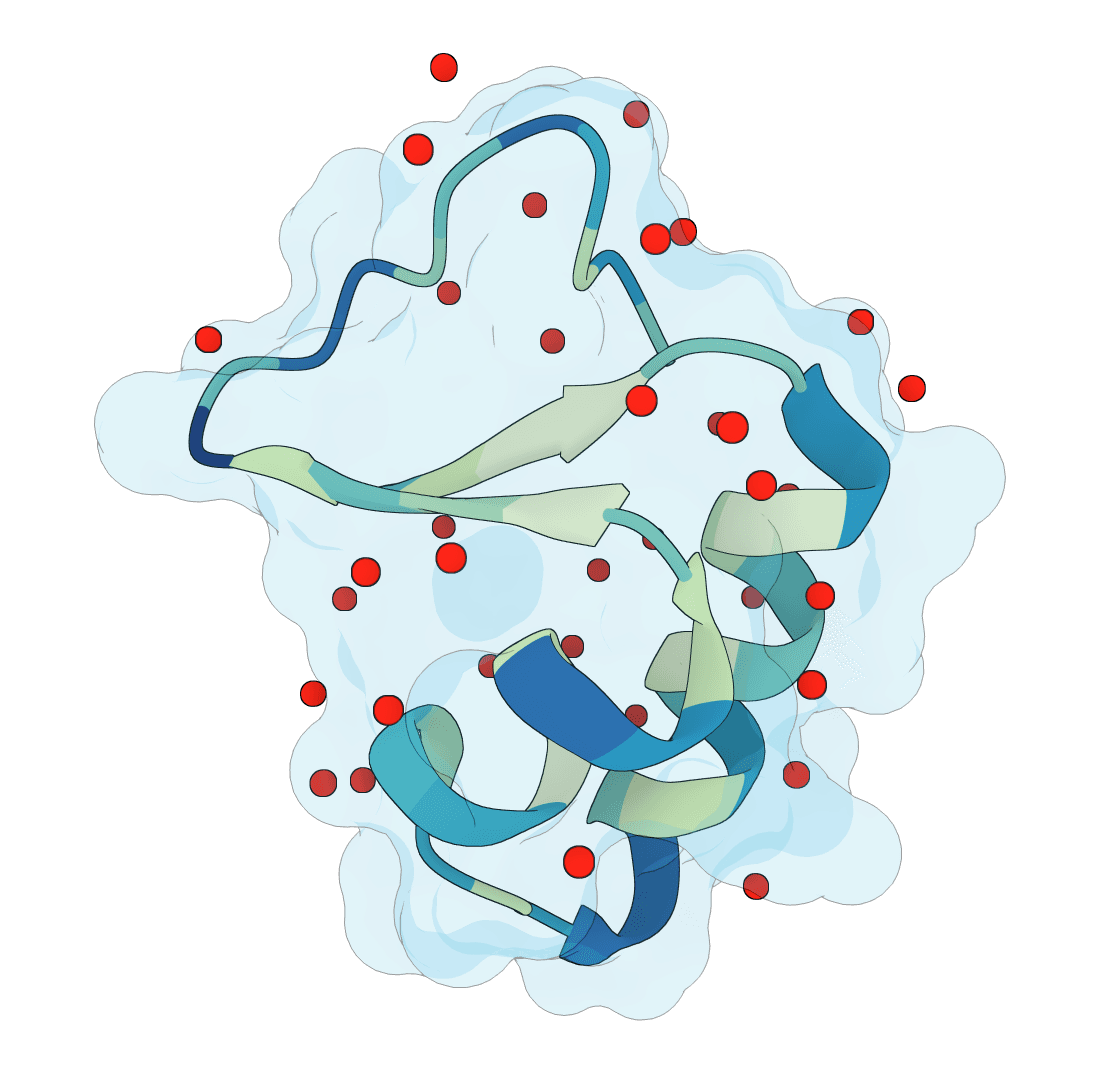
Predict protein hydration sites from a structure using a diffusion model with ESM features and a confidence-filtering head.
Compute 200+ RDKit molecular descriptors, drug-likeness rule violations, and structural fingerprints for QSAR, virtual screening, and ML workflows
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.
Predict protein thermostability changes (ΔΔG) for point mutations using a graph neural network. Enables computational saturation mutagenesis screening to identify stabilizing mutations.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
Predict protein solubility from amino acid sequence using the University of Manchester Protein-Sol method.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Predict protein hydration sites from a structure using a diffusion model with ESM features and a confidence-filtering head.
Compute 200+ RDKit molecular descriptors, drug-likeness rule violations, and structural fingerprints for QSAR, virtual screening, and ML workflows
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Designed nanobody stability shortlist
Stability assessment
This tool predicts protein stability from amino acid sequences using validated physicochemical methods. The stability score combines multiple peer-reviewed predictors to estimate how stable a protein is likely to be under physiological conditions.
The final stability score (0-100) formula combines five predictors, each weighted based on its estimated contribution to protein stability:
| Score | Category | Interpretation |
|---|---|---|
| 80-100 | Very stable | Excellent stability predicted |
| 60-79 | Stable | Good stability under normal conditions |
| 40-59 | Moderately stable | May require optimization |
| 20-39 | Unstable | Likely stability issues |
| 0-19 | Very unstable | Significant stability concerns |
Based on Guruprasad et al. (1990), the Instability Index estimates protein stability based on dipeptide composition. Statistical analysis revealed that certain dipeptide pairs occur at significantly different frequencies in stable versus unstable proteins.
This is the most heavily weighted factor because it's based on large-scale statistical analysis of protein stability.
The Aliphatic Index (Ikai (1980)) represents the relative volume occupied by aliphatic side chains (Ala, Val, Ile, Leu). Proteins with higher aliphatic content tend to be more stable at elevated temperatures.
This metric is valuable for thermostability engineering because aliphatic residues contribute to hydrophobic core packing.
It's calculated with the following formula:
where is mole percent.
Typical range of values is between 0 and 120, with values greater than 80 meaning good thermostability.
GRAVY (Kyte & Doolittle (1982)) measures the average hydrophobicity by summing hydropathy values for each amino acid and dividing by sequence length. Balanced hydrophobicity is crucial for proper folding and stability.
Extreme GRAVY values often indicate potential stability or solubility issues requiring experimental validation.
The Flexibility Index (Vihinen et al. (1994)) predicts local and global protein flexibility based on normalized amino acid composition. Rigid proteins typically exhibit better stability than highly flexible ones.
Reduced flexibility often correlates with improved stability because rigid structures are less prone to unfolding.
Charge density measures the proportion of charged residues (Lys, Arg, His, Asp, Glu) relative to sequence length. High charge density can lead to electrostatic repulsion and reduced stability.
Lower charge density generally correlates with better stability, though context-dependent (e.g., pH, ionic strength).
Step 1: Screen sequences - Calculate stability scores for all candidates
Step 2: Analyze components - Review individual metrics (II, AI, GRAVY, etc.)
Step 3: Identify issues - Look for high instability index, low aliphatic index, extreme GRAVY
Step 4: Engineer improvements - Use metrics to guide rational mutagenesis
Step 5: Validate experimentally - Confirm predictions with thermal shift assays, DSC, or stability studies