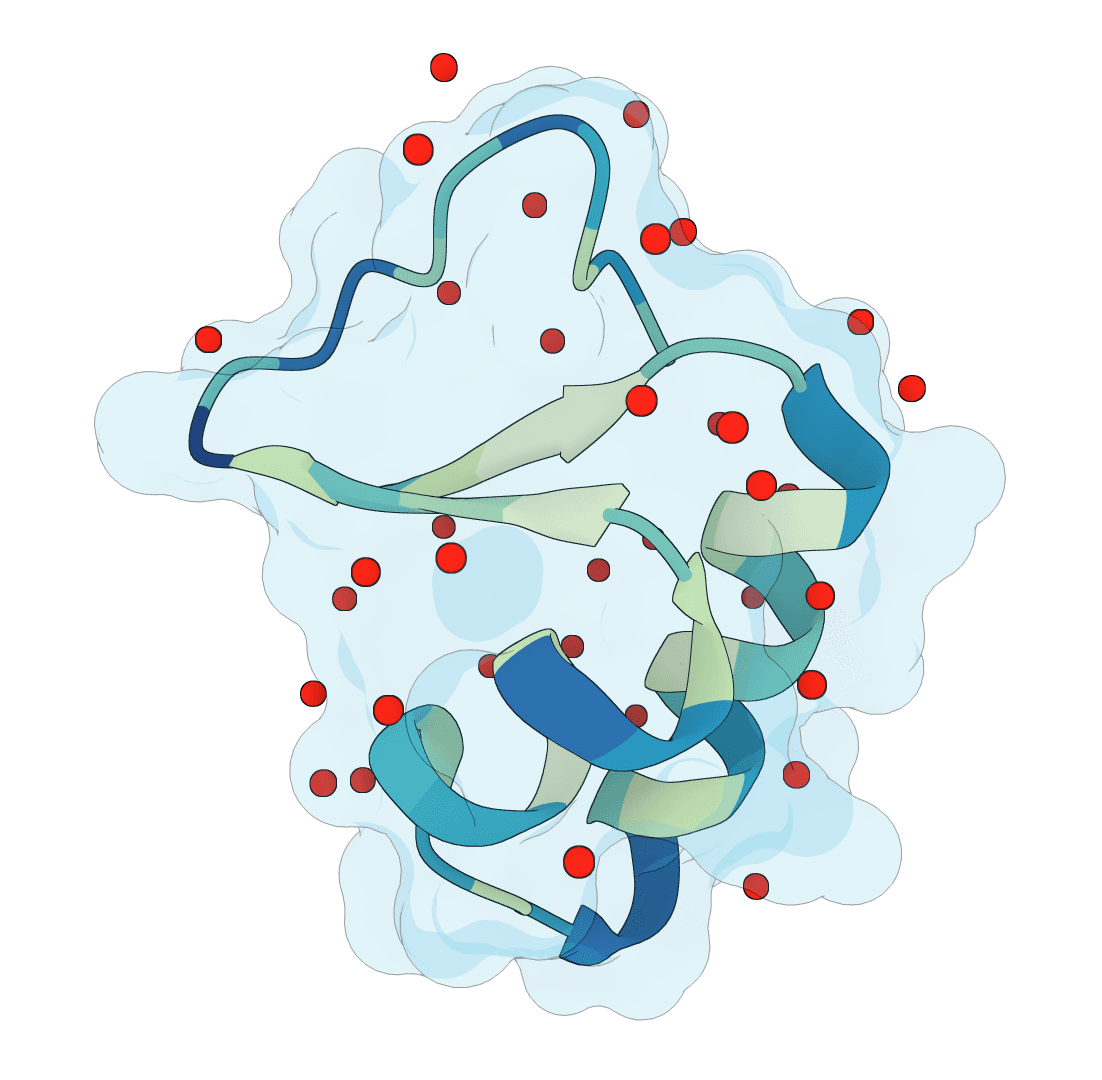
SuperWater predicts ordered water molecule positions around protein structures. It uses a generative model to sample candidate hydration sites, then filters and clusters those candidates to recover likely crystallographic and interface waters from a single structure.
Published in Communications Chemistry in December 2025, SuperWater was introduced as a score-based diffusion framework with equivariant graph neural networks and ESM-derived features. In the reported benchmarks, it outperformed HydraProt and GalaxyWater-CNN across much of the precision-coverage range for protein surface waters as well as protein-protein and protein-ligand interface waters.
ProteinIQ provides browser-based access to SuperWater on hosted compute, so hydration-site prediction can be run from an uploaded structure or an RCSB entry without local setup.
| Input | Description |
|---|---|
Protein Structure | One protein structure in PDB, ENT, CIF, mmCIF, or PDBx format, or a structure fetched from RCSB. The file must contain protein atoms. Maximum file size is 50 MB. |
| Setting | Description |
|---|---|
Water ratio | Approximate number of sampled water candidates per residue. Range 1-10, default 8. Higher values increase coverage but also runtime and memory use. Larger structures may automatically route to higher GPU tiers, and combinations beyond the supported GPU ceiling are blocked before launch. |
Confidence cutoff | Minimum confidence score required to keep a predicted hydration site. Range 0.02-0.5, default 0.1. Higher values improve precision but return fewer waters. |
Inference steps | Reverse-diffusion steps used during sampling. 10 is faster, 20 is the default, and 30 may improve quality at the cost of runtime. |
ProteinIQ returns a 3D viewer, a tabular summary, and downloadable result files.
| Output | Description |
|---|---|
Viewer | Interactive structure view with predicted water coordinates included in the returned files. |
Data table | Summary table for the processed structure and prediction settings. |
Files | Downloadable structure and result files, including the combined structure with predicted waters and auxiliary confidence outputs. |
| Column | Description |
|---|---|
Structure | Input structure name or identifier |
Predicted waters | Number of hydration sites retained after confidence filtering and clustering |
Sampled candidates | Number of candidate positions generated before final filtering |
Max confidence | Highest confidence assigned to any retained water site |
Mean confidence | Average confidence across retained water sites |
Water ratio | Water sampling multiplier used for the run |
Confidence cutoff | Acceptance threshold used to keep predicted waters |
Input source | Whether the structure came from file upload or RCSB fetch |
SuperWater does not score a fixed 3D voxel grid the way earlier hydration predictors do. Instead, it learns the gradient of the water-position distribution around a protein and uses that learned score field to refine randomly initialized water coordinates through reverse diffusion.
The architecture reported in the paper combines a score-based diffusion model with equivariant graph neural networks, which preserves geometric consistency under rotation. The published method also incorporates ESM features to encode sequence-derived context around residues. After sampling, a separate confidence model removes low-probability positions, and a clustering step consolidates nearby candidates into final hydration sites.
The benchmark protocol described in the paper sampled an initial number of candidates proportional to protein length, then traced different precision-coverage tradeoffs by varying the internal confidence threshold (cap). On an independent test set of 1,709 crystal structures, SuperWater defined the best overall precision-coverage frontier among the compared methods across many operating points.
SuperWater predictions represent likely ordered water sites, not every transient solvent molecule around the protein. High-confidence predictions are more likely to correspond to persistent hydration sites that recur in experimental structures or stabilize interfaces.
| Confidence pattern | Interpretation |
|---|---|
High Max confidence with moderate-to-high Predicted waters | Strong evidence for a structured hydration pattern around the input fold or interface |
Low Confidence cutoff and many returned waters | Broader coverage, useful for exploratory analysis but more likely to include false positives |
Higher Confidence cutoff and fewer returned waters | More conservative set of hydration sites, typically better for prioritizing inspection |
Large gap between Sampled candidates and Predicted waters | Many sampled positions were rejected during filtering, indicating a stricter or more selective final result |
The paper reports mean absolute deviation of approximately 0.3 ± 0.06 Å at cap = 0.5 for matched predictions, which indicates that true-positive sites can be placed with sub-angstrom accuracy. That figure should be interpreted as benchmarked localization accuracy against crystallographic waters, not as a guarantee for every structure.
SuperWater predicts positions from a static protein structure. It does not explicitly model long-timescale solvent dynamics, alternate conformations, protonation-state uncertainty, or experimental conditions such as crystal packing, buffer composition, or ligand occupancy.
Like other hydration-site predictors, it is biased toward ordered waters that are recoverable from structural datasets. Disordered, low-occupancy, or rapidly exchanging solvent molecules may be absent from the output even when they are biologically relevant.
Prediction counts also depend directly on Water ratio and Confidence cutoff. A denser sampling strategy can improve recall, but it does not by itself validate the biological importance of every returned site.
ProteinIQ accepts large structures by routing them across multiple GPU tiers, but there is still a hard ceiling for very large residue count × water ratio combinations. If a submission exceeds that supported range, lower Water ratio or use a smaller structure.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
LocScale performs physics-informed local sharpening of cryo-EM density maps using half-maps or full MRC/MAP volumes, with optional mask and reference-map inputs.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.
Predict protein solubility from amino acid sequence using the University of Manchester Protein-Sol method.
SuperWater predicts ordered water molecule positions around protein structures. It uses a generative model to sample candidate hydration sites, then filters and clusters those candidates to recover likely crystallographic and interface waters from a single structure.
Published in Communications Chemistry in December 2025, SuperWater was introduced as a score-based diffusion framework with equivariant graph neural networks and ESM-derived features. In the reported benchmarks, it outperformed HydraProt and GalaxyWater-CNN across much of the precision-coverage range for protein surface waters as well as protein-protein and protein-ligand interface waters.
ProteinIQ provides browser-based access to SuperWater on hosted compute, so hydration-site prediction can be run from an uploaded structure or an RCSB entry without local setup.
| Input | Description |
|---|---|
Protein Structure | One protein structure in PDB, ENT, CIF, mmCIF, or PDBx format, or a structure fetched from RCSB. The file must contain protein atoms. Maximum file size is 50 MB. |
| Setting | Description |
|---|---|
Water ratio | Approximate number of sampled water candidates per residue. Range 1-10, default 8. Higher values increase coverage but also runtime and memory use. Larger structures may automatically route to higher GPU tiers, and combinations beyond the supported GPU ceiling are blocked before launch. |
Confidence cutoff | Minimum confidence score required to keep a predicted hydration site. Range 0.02-0.5, default 0.1. Higher values improve precision but return fewer waters. |
Inference steps | Reverse-diffusion steps used during sampling. 10 is faster, 20 is the default, and 30 may improve quality at the cost of runtime. |
ProteinIQ returns a 3D viewer, a tabular summary, and downloadable result files.
| Output | Description |
|---|---|
Viewer | Interactive structure view with predicted water coordinates included in the returned files. |
Data table | Summary table for the processed structure and prediction settings. |
Files | Downloadable structure and result files, including the combined structure with predicted waters and auxiliary confidence outputs. |
| Column | Description |
|---|---|
Structure | Input structure name or identifier |
Predicted waters | Number of hydration sites retained after confidence filtering and clustering |
Sampled candidates | Number of candidate positions generated before final filtering |
Max confidence | Highest confidence assigned to any retained water site |
Mean confidence | Average confidence across retained water sites |
Water ratio | Water sampling multiplier used for the run |
Confidence cutoff | Acceptance threshold used to keep predicted waters |
Input source | Whether the structure came from file upload or RCSB fetch |
SuperWater does not score a fixed 3D voxel grid the way earlier hydration predictors do. Instead, it learns the gradient of the water-position distribution around a protein and uses that learned score field to refine randomly initialized water coordinates through reverse diffusion.
The architecture reported in the paper combines a score-based diffusion model with equivariant graph neural networks, which preserves geometric consistency under rotation. The published method also incorporates ESM features to encode sequence-derived context around residues. After sampling, a separate confidence model removes low-probability positions, and a clustering step consolidates nearby candidates into final hydration sites.
The benchmark protocol described in the paper sampled an initial number of candidates proportional to protein length, then traced different precision-coverage tradeoffs by varying the internal confidence threshold (cap). On an independent test set of 1,709 crystal structures, SuperWater defined the best overall precision-coverage frontier among the compared methods across many operating points.
SuperWater predictions represent likely ordered water sites, not every transient solvent molecule around the protein. High-confidence predictions are more likely to correspond to persistent hydration sites that recur in experimental structures or stabilize interfaces.
| Confidence pattern | Interpretation |
|---|---|
High Max confidence with moderate-to-high Predicted waters | Strong evidence for a structured hydration pattern around the input fold or interface |
Low Confidence cutoff and many returned waters | Broader coverage, useful for exploratory analysis but more likely to include false positives |
Higher Confidence cutoff and fewer returned waters | More conservative set of hydration sites, typically better for prioritizing inspection |
Large gap between Sampled candidates and Predicted waters | Many sampled positions were rejected during filtering, indicating a stricter or more selective final result |
The paper reports mean absolute deviation of approximately 0.3 ± 0.06 Å at cap = 0.5 for matched predictions, which indicates that true-positive sites can be placed with sub-angstrom accuracy. That figure should be interpreted as benchmarked localization accuracy against crystallographic waters, not as a guarantee for every structure.
SuperWater predicts positions from a static protein structure. It does not explicitly model long-timescale solvent dynamics, alternate conformations, protonation-state uncertainty, or experimental conditions such as crystal packing, buffer composition, or ligand occupancy.
Like other hydration-site predictors, it is biased toward ordered waters that are recoverable from structural datasets. Disordered, low-occupancy, or rapidly exchanging solvent molecules may be absent from the output even when they are biologically relevant.
Prediction counts also depend directly on Water ratio and Confidence cutoff. A denser sampling strategy can improve recall, but it does not by itself validate the biological importance of every returned site.
ProteinIQ accepts large structures by routing them across multiple GPU tiers, but there is still a hard ceiling for very large residue count × water ratio combinations. If a submission exceeds that supported range, lower Water ratio or use a smaller structure.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
LocScale performs physics-informed local sharpening of cryo-EM density maps using half-maps or full MRC/MAP volumes, with optional mask and reference-map inputs.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.
Predict protein solubility from amino acid sequence using the University of Manchester Protein-Sol method.