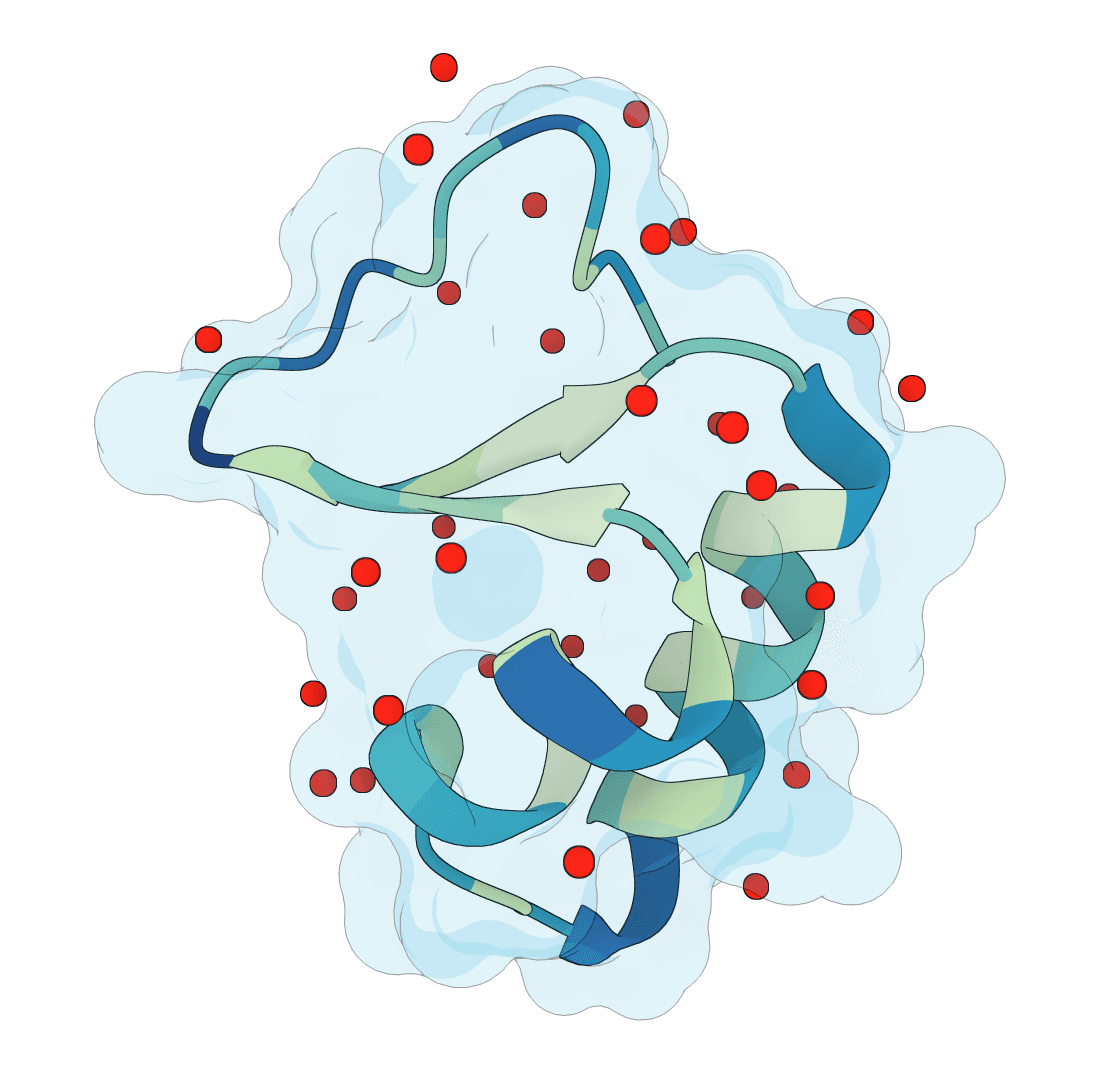
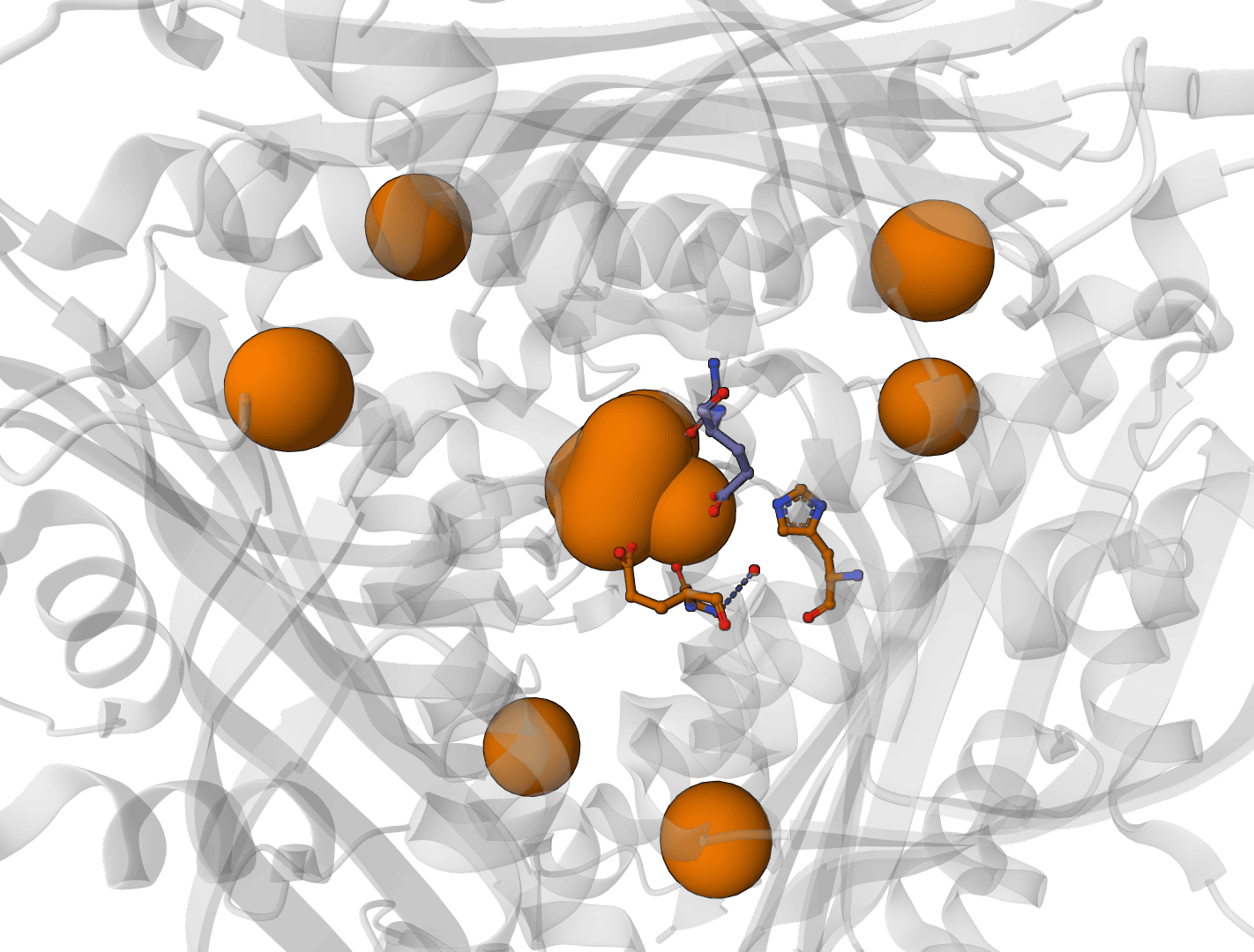AllMetal3D predicts metal binding sites in protein structures, including where a site is likely to occur, the most likely native identity class, and the coordination geometry. Its identity classifier reports the same classes available in the original project: Alkali, MG, CA, ZN, NonZNTM, and NoMetal. A companion model, Water3D, predicts likely water binding positions in the same framework.
Developed by Simon Duerr and Ursula Roethlisberger at EPFL's Laboratory of Computational Chemistry and Biochemistry, AllMetal3D addresses a persistent blind spot in structure prediction: deposited PDB structures frequently have missing or misassigned metal ions, and apo structures lack metals entirely. AllMetal3D benchmarks favorably against MetalSiteHunter, AlphaFold3, MIC, MIB2, and MetalHawk.
ProteinIQ runs AllMetal3D and Water3D on cloud GPU infrastructure—no software installation, no Python environment.
Settings
Output
Results for AllMetal3D are tabulated by site:
The detailed table also includes raw per-class probability columns for both identity and geometry, so you can inspect the full native distribution instead of only the top-ranked class.
Downloadable outputs include the predicted PDB (with metal coordinates appended) and CUBE-format electron density files for visualization.
AllMetal3D uses a fully convolutional 3D CNN trained on metal-containing structures from the PDB. The network processes a volumetric representation of the local protein environment centered on candidate positions. The architecture consists of five convolutional layers with 8, 60, 100, 80, and 30 channels, with leaky ReLU activation (slope 0.2) and max-pooling after the second layer. Processed features condense into a 1,280-dimensional fingerprint that feeds into separate fully-connected heads for identity and geometry classification.
Prediction follows a two-stage pipeline. AllMetal3Dloc first identifies candidate positions by scanning the structure and scoring each position for the likelihood of containing any metal. Positions above the probability threshold are then passed to the classification network, which outputs the most probable metal identity and coordination geometry.
Water3D applies the same 3D CNN framework trained specifically on crystallographic water positions, identifying ordered water molecules that are consistently present across multiple crystal structures of related proteins.
Interpreting results
Identity classification accuracy varies substantially by metal type. Zinc sites are predicted most reliably given the abundance of Zn-containing structures in training data. Magnesium and sodium are more challenging due to their broad and irregular binding motifs.
Coordination geometry
The geometry classifier performs well for tetrahedral and octahedral arrangements, which dominate in natural proteins. Other geometries (square planar, trigonal bipyramidal, etc.) are classified less accurately and should be treated with caution.
Water3D sites
Water predictions reflect ordered, structurally conserved water positions. Loosely bound or disordered waters—which appear at low occupancy in crystal structures—will generally fall below the probability threshold.
Limitations
Metal selectivity depends partly on cellular context (compartmentalization, metal availability during protein synthesis) that cannot be inferred from structure alone. A site predicted as Mg²⁺ may be occupied by other divalent ions in different cellular environments.
Median positional error for metal site location is approximately 0.5 Å. This is sufficient for identifying which residues coordinate the metal, but marginal for distinguishing subtle geometry differences. Vacancy prediction—identifying sites that are empty in a given structure but could be occupied—was not reliable and is not reported.
Performance on Mg²⁺ and Na⁺/K⁺ is meaningfully lower than for transition metals, reflecting sparser training data and the tendency of these ions to adopt irregular or water-mediated coordination environments.