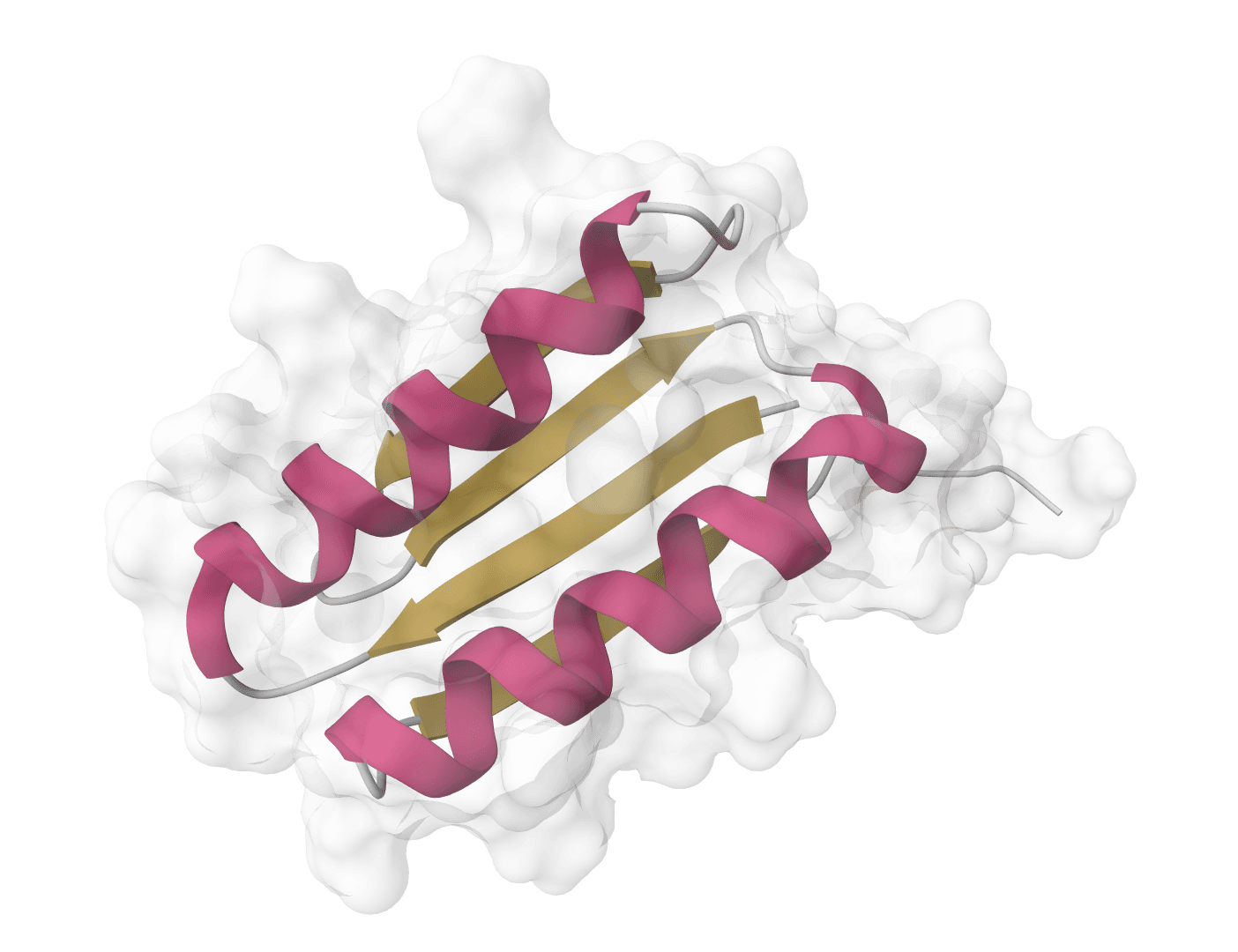
Assign helices, sheets, loops, and hydrogen bonds from protein structure coordinates. Learn more
Run
Output
Configure inputs to begin
Set options on the left, then click “Analyze”.
Assign helices, sheets, loops, and hydrogen bonds from protein structure coordinates. Learn more
Configure inputs to begin
Set options on the left, then click “Analyze”.
Assign helices, sheets, loops, and hydrogen bonds from protein structure coordinates. Learn more
Configure inputs to begin
Set options on the left, then click “Analyze”.
DSSP (Dictionary of Secondary Structure of Proteins) is the gold standard algorithm for assigning protein secondary structure from atomic coordinates. Developed by Kabsch and Sander in 1983, DSSP analyzes hydrogen bonding patterns in protein backbones to classify each residue as helix, sheet, turn, bend, or loop.
Unlike methods that rely on annotated HELIX and SHEET records in PDB files, DSSP calculates secondary structure directly from atomic positions. This makes it ideal for analyzing predicted structures from AlphaFold, ESMFold, or Boltz-2, which typically lack these annotations.
DSSP remains the most widely used secondary structure assignment tool in structural biology, serving as the reference standard against which other methods are validated.
DSSP identifies backbone hydrogen bonds using the Kabsch-Sander electrostatic model. The algorithm assumes partial charges of on the carbonyl oxygen and on the amide hydrogen.
The hydrogen bond energy between a donor N-H and acceptor C=O is calculated as:
where , , , and are distances (in Ångströms) between the respective atom pairs. A hydrogen bond exists when kcal/mol.
DSSP assigns eight structure types based on hydrogen bonding patterns:
| Code | Structure | Description |
|---|---|---|
| H | α-helix | 4-helix with i→i+4 hydrogen bonds |
| G | 3₁₀-helix | 3-helix with i→i+3 hydrogen bonds |
| I | π-helix | 5-helix with i→i+5 hydrogen bonds |
| E | β-strand | Extended strand in β-sheet |
| B | β-bridge | Isolated β-bridge (single residue) |
| T | Turn | Hydrogen-bonded turn |
| S | Bend | Backbone angle > 70° |
| (space) | Loop | No defined structure |
Helices form when consecutive n-turns overlap. An n-turn exists when residue i+n donates a hydrogen bond to residue i. Two consecutive 4-turns create an α-helix (H), two consecutive 3-turns create a 3₁₀-helix (G), and two consecutive 5-turns create a π-helix (I).
β-strands are identified through ladder patterns where residues form parallel or antiparallel hydrogen-bonded bridges. A single bridge is classified as B, while consecutive bridges form extended strands (E).
Bends are assigned when the angle between Cα positions at i-2, i, and i+2 exceeds 70°, indicating a sharp change in backbone direction.
Upload PDB files or fetch structures using 4-letter PDB IDs. DSSP requires complete backbone atoms (N, CA, C, O) for each residue. Missing backbone atoms will cause those residues to be skipped.
Summary — Provides per-chain percentages of helix, sheet, and loop content. This condensed view is useful for comparing overall structural composition across multiple proteins.
Per residue — Shows DSSP codes for every amino acid along with hydrogen bond partners. Use this detailed output to identify specific structural elements or analyze local folding patterns.
The summary table shows structural composition for each chain:
Each row represents one amino acid with its assigned structure code and hydrogen bond partners:
Hydrogen bond partners help identify interaction patterns. For example, residues in an α-helix show i→i+4 bonding patterns.
α-helices (H) are the most common secondary structure, typically comprising 30-35% of globular proteins. They form stable, right-handed helical structures with 3.6 residues per turn.
β-strands (E) usually represent 20-25% of protein structure. They combine with other strands to form β-sheets, which can be parallel, antiparallel, or mixed.
Loops and coils (space) connect structured elements and often contain functionally important binding sites or catalytic residues. They typically make up 15-25% of the structure.
3₁₀-helices (G) are shorter and tighter than α-helices, often appearing at helix ends or in tight turns. π-helices (I) are rare, comprising less than 1% of residues.
DSSP requires complete backbone atoms (N, CA, C, O) for accurate assignment. Structures with missing atoms will have incomplete results. Use PDB Fixer to add missing atoms before running DSSP.
The algorithm is sensitive to coordinate precision. Low-resolution structures or poorly refined models may produce unreliable assignments, particularly for borderline cases like short helices or isolated bridges.
DSSP cannot distinguish between static and dynamic secondary structure. Flexible regions that transiently adopt helical or extended conformations are assigned based on their conformation in the input structure.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.

Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Calculate pairwise RMSD matrices for PDB structure ensembles with pyRMSD, including the condensed matrix and source statistics files.
Calculate the radius of gyration (Rg) for protein structures from PDB files. Supports multiple chains and atom selection options.
Calculate RMSD between protein structures with optimal rigid-body alignment, explicit atom correspondence, coverage diagnostics, per-residue results, and fitted coordinate files.
Calculate Solvent Accessible Surface Area (SASA) for protein structures using the Shrake-Rupley algorithm.
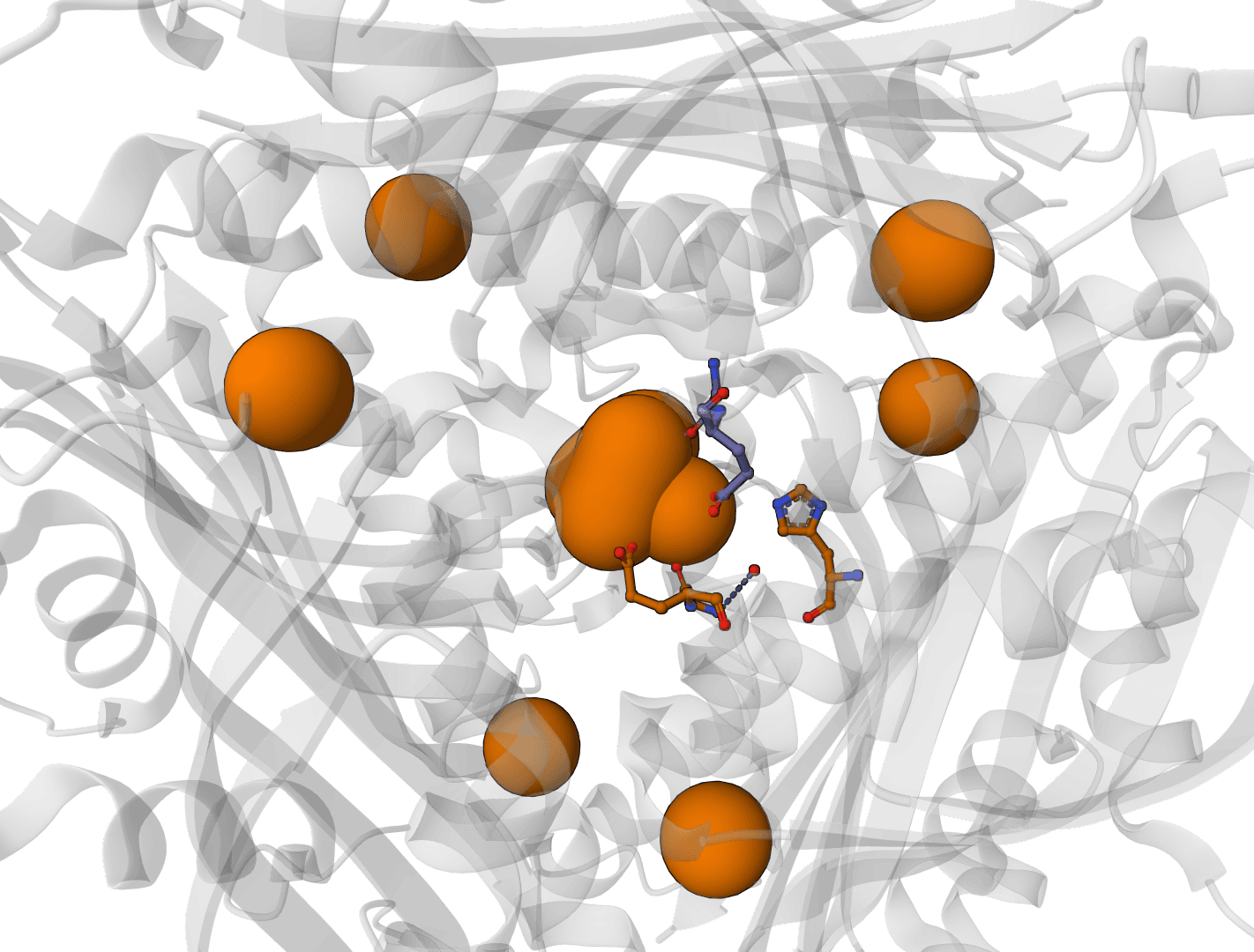
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
LocScale performs physics-informed local sharpening of cryo-EM density maps using half-maps or full MRC/MAP volumes, with optional mask and reference-map inputs.
DSSP (Dictionary of Secondary Structure of Proteins) is the gold standard algorithm for assigning protein secondary structure from atomic coordinates. Developed by Kabsch and Sander in 1983, DSSP analyzes hydrogen bonding patterns in protein backbones to classify each residue as helix, sheet, turn, bend, or loop.
Unlike methods that rely on annotated HELIX and SHEET records in PDB files, DSSP calculates secondary structure directly from atomic positions. This makes it ideal for analyzing predicted structures from AlphaFold, ESMFold, or Boltz-2, which typically lack these annotations.
DSSP remains the most widely used secondary structure assignment tool in structural biology, serving as the reference standard against which other methods are validated.
DSSP identifies backbone hydrogen bonds using the Kabsch-Sander electrostatic model. The algorithm assumes partial charges of on the carbonyl oxygen and on the amide hydrogen.
The hydrogen bond energy between a donor N-H and acceptor C=O is calculated as:
where , , , and are distances (in Ångströms) between the respective atom pairs. A hydrogen bond exists when kcal/mol.
DSSP assigns eight structure types based on hydrogen bonding patterns:
| Code | Structure | Description |
|---|---|---|
| H | α-helix | 4-helix with i→i+4 hydrogen bonds |
| G | 3₁₀-helix | 3-helix with i→i+3 hydrogen bonds |
| I | π-helix | 5-helix with i→i+5 hydrogen bonds |
| E | β-strand | Extended strand in β-sheet |
| B | β-bridge | Isolated β-bridge (single residue) |
| T | Turn | Hydrogen-bonded turn |
| S | Bend | Backbone angle > 70° |
| (space) | Loop | No defined structure |
Helices form when consecutive n-turns overlap. An n-turn exists when residue i+n donates a hydrogen bond to residue i. Two consecutive 4-turns create an α-helix (H), two consecutive 3-turns create a 3₁₀-helix (G), and two consecutive 5-turns create a π-helix (I).
β-strands are identified through ladder patterns where residues form parallel or antiparallel hydrogen-bonded bridges. A single bridge is classified as B, while consecutive bridges form extended strands (E).
Bends are assigned when the angle between Cα positions at i-2, i, and i+2 exceeds 70°, indicating a sharp change in backbone direction.
Upload PDB files or fetch structures using 4-letter PDB IDs. DSSP requires complete backbone atoms (N, CA, C, O) for each residue. Missing backbone atoms will cause those residues to be skipped.
Summary — Provides per-chain percentages of helix, sheet, and loop content. This condensed view is useful for comparing overall structural composition across multiple proteins.
Per residue — Shows DSSP codes for every amino acid along with hydrogen bond partners. Use this detailed output to identify specific structural elements or analyze local folding patterns.
The summary table shows structural composition for each chain:
Each row represents one amino acid with its assigned structure code and hydrogen bond partners:
Hydrogen bond partners help identify interaction patterns. For example, residues in an α-helix show i→i+4 bonding patterns.
α-helices (H) are the most common secondary structure, typically comprising 30-35% of globular proteins. They form stable, right-handed helical structures with 3.6 residues per turn.
β-strands (E) usually represent 20-25% of protein structure. They combine with other strands to form β-sheets, which can be parallel, antiparallel, or mixed.
Loops and coils (space) connect structured elements and often contain functionally important binding sites or catalytic residues. They typically make up 15-25% of the structure.
3₁₀-helices (G) are shorter and tighter than α-helices, often appearing at helix ends or in tight turns. π-helices (I) are rare, comprising less than 1% of residues.
DSSP requires complete backbone atoms (N, CA, C, O) for accurate assignment. Structures with missing atoms will have incomplete results. Use PDB Fixer to add missing atoms before running DSSP.
The algorithm is sensitive to coordinate precision. Low-resolution structures or poorly refined models may produce unreliable assignments, particularly for borderline cases like short helices or isolated bridges.
DSSP cannot distinguish between static and dynamic secondary structure. Flexible regions that transiently adopt helical or extended conformations are assigned based on their conformation in the input structure.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Calculate pairwise RMSD matrices for PDB structure ensembles with pyRMSD, including the condensed matrix and source statistics files.
Calculate the radius of gyration (Rg) for protein structures from PDB files. Supports multiple chains and atom selection options.
Calculate RMSD between protein structures with optimal rigid-body alignment, explicit atom correspondence, coverage diagnostics, per-residue results, and fitted coordinate files.
Calculate Solvent Accessible Surface Area (SASA) for protein structures using the Shrake-Rupley algorithm.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
LocScale performs physics-informed local sharpening of cryo-EM density maps using half-maps or full MRC/MAP volumes, with optional mask and reference-map inputs.