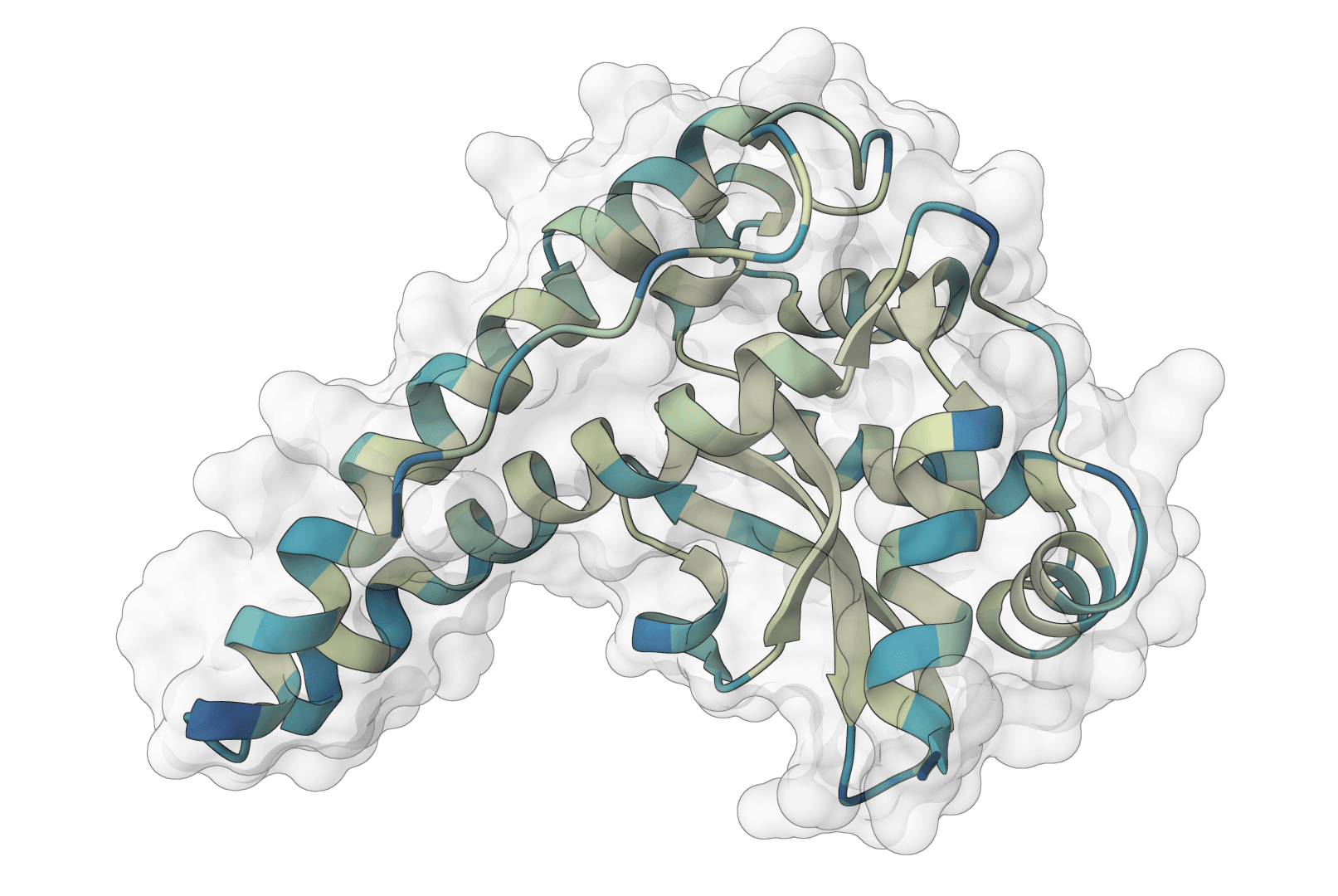
Run
50 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Human ubiquitin — MSA-assisted monomer (76 aa)
GCN4 leucine zipper — homodimer (33 aa × 2)
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Human ubiquitin — MSA-assisted monomer (76 aa)
GCN4 leucine zipper — homodimer (33 aa × 2)
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Human ubiquitin — MSA-assisted monomer (76 aa)
GCN4 leucine zipper — homodimer (33 aa × 2)
AlphaFold2 is the protein structure prediction model that revolutionized structural biology when DeepMind released it in 2021. By analyzing evolutionary relationships encoded in multiple sequence alignments (MSA), AlphaFold2 predicts 3D protein structures with accuracy rivaling experimental methods.
ProteinIQ runs ColabFold with two access paths. Guest and free-plan runs default to single-sequence inference with no database search. Paid plans can generate MSAs through MMseqs2, including the broad UniRef+Environmental mode used for the strongest evolutionary context.
AlphaFold2 supports both single-chain (monomer) and multi-chain (multimer) predictions. For complex structures involving ligands, DNA, or RNA, consider newer models like Boltz-2 or Chai-1 which extend beyond protein-only predictions.
Provide one or more protein sequences to generate ranked AlphaFold2 monomer or multimer structures online. Free runs use single-sequence mode, while paid plans can add MMseqs2 database searches for MSA-assisted prediction. Results include PDB structures, pLDDT, PAE, pTM, and ipTM metrics where applicable.
AlphaFold2 extracts coevolutionary information from MSAs—when two residue positions consistently change together across evolution, they're likely in spatial contact. The model transforms this evolutionary signal into accurate 3D coordinates.
The core innovation is the Evoformer module, a neural network that processes both sequence alignments and pairwise residue relationships simultaneously. Through 48 repeated blocks, information flows between these two representations, building increasingly refined structural features.
The MSA representation captures which residues are evolutionarily constrained and how they co-vary. The pair representation accumulates spatial relationship predictions between every residue pair.
After Evoformer processing, the structure module converts learned features into 3D atomic coordinates. It predicts backbone frames (position and orientation for each residue) and refines side-chain positions.
The prediction process includes recycling—feeding outputs back through the network for iterative refinement. Each recycle step improves the structure, particularly for challenging regions where initial predictions may be uncertain.
AlphaFold2-Multimer extends the base model to protein complexes. It uses paired MSAs that capture inter-chain coevolution, helping the model predict how chains interact. The model automatically detects if your input contains multiple chains and switches to multimer mode.
Provide protein sequences via text input (raw sequence or FASTA format), file upload (.fasta, .fa, .txt, .a3m), or PDB ID fetch from RCSB.
For multimer predictions, add multiple chains as separate inputs. Chain IDs are assigned automatically (A, B, C...). You can predict up to 10 chains in a single job.
3; increase to 6–12 for challenging predictions.Auto selects the appropriate variant based on your input—ptm for single chains, multimer v3 for complexes. Force a specific type only if you have a reason (e.g., benchmarking).MSA quality is the single most important factor for prediction accuracy. Deeper alignments with more homologous sequences generally produce better predictions.
No MSA disables sequence search and makes AlphaFold2 behave like a single-sequence model, similar to ESMFold.UniRef+Environmental searches the broadest set, including metagenomic sequences, and is the paid default. UniRef only is faster but may miss distant homologs. Remote MSA modes use twice the base credits shown for single-sequence mode.Unpaired+Paired combines chain-specific and cross-chain coevolution signals. Paired only uses only sequences where both chains appear together. Unpaired only treats chains independently.AlphaFold2 generates multiple ranked predictions, each with confidence metrics that help you assess reliability.
The predicted Local Distance Difference Test (pLDDT) estimates how accurate each residue's position is, stored in the B-factor column of output PDB files.
| pLDDT | Confidence | Interpretation |
|---|---|---|
| > 90 | Very high | High confidence in backbone and side chains |
| 70–90 | Good | Backbone likely correct; side chains may vary |
| 50–70 | Low | Structural uncertainty; interpret with caution |
| < 50 | Very low | Often indicates disorder or flexibility |
Residues with pLDDT below 50 frequently represent intrinsically disordered regions (IDRs) that lack fixed structure rather than prediction failures.
The predicted Template Modeling score (pTM) estimates overall structural accuracy, ranging from 0 to 1. Values above 0.7 typically indicate reliable predictions.
For multimers, the interface pTM (ipTM) specifically assesses inter-chain interface quality. The combined ranking score weights both metrics: ranking_score = 0.8 × ipTM + 0.2 × pTM. Combined scores above 0.62 suggest reliable complex predictions.
The Predicted Aligned Error matrix shows expected position errors between all residue pairs. Low PAE values (blue in visualizations) indicate confident relative positioning; high values (red) suggest uncertainty.
PAE is particularly useful for:
AlphaFold2 ranks predictions by confidence scores. We recommend examining the top 2–3 structures rather than only the #1 result—especially for flexible proteins or novel folds where ranking may be uncertain.
| Feature | AlphaFold2 | ESMFold | Boltz-2 | Chai-1 |
|---|---|---|---|---|
| Speed | Fast without MSA; moderate with MSA | Very fast (~seconds) | Moderate | Moderate |
| MSA required | Optional; paid modes add database search | No | Optional | Optional |
| Accuracy | Depends on target and MSA mode | Good (~0.95 TM) | Excellent | Excellent |
| Multimer support | Yes | Yes | Yes | Yes |
| Ligand prediction | No | No | Yes | Yes |
| DNA/RNA support | No | No | Yes | Yes |
Use AlphaFold2 when the established protein-only prediction workflow is needed. MSA-assisted paid modes are especially valuable for natural proteins with detectable evolutionary homologs; free single-sequence mode is faster but may be less accurate on difficult targets.
Use ESMFold when speed matters most or you're working with designed proteins that lack evolutionary relatives. ESMFold skips MSA generation entirely.
Use MiniFold when a fast single-sequence monomer predictor is enough and AlphaFold2's MSA search would be unnecessary overhead.
Use Boltz-2 or Chai-1 when you need to predict complexes involving small molecules, DNA, or RNA, or when you want binding affinity estimates alongside structure predictions.
Use OpenFold 3 or Protenix when you want open-source implementations of AlphaFold3-like capabilities for multi-molecule complexes.
AlphaFold2 requires evolutionarily related sequences for optimal performance. Predictions for de novo designed proteins or sequences without detectable homologs will have lower confidence.
The model predicts static structures and may not capture conformational changes, allosteric states, or dynamics. Regions with pLDDT below 50 often represent genuine flexibility rather than prediction errors.
AlphaFold2 cannot predict ligand binding, post-translational modifications, or the effects of small molecules on structure. For these applications, use newer models like Boltz-2.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
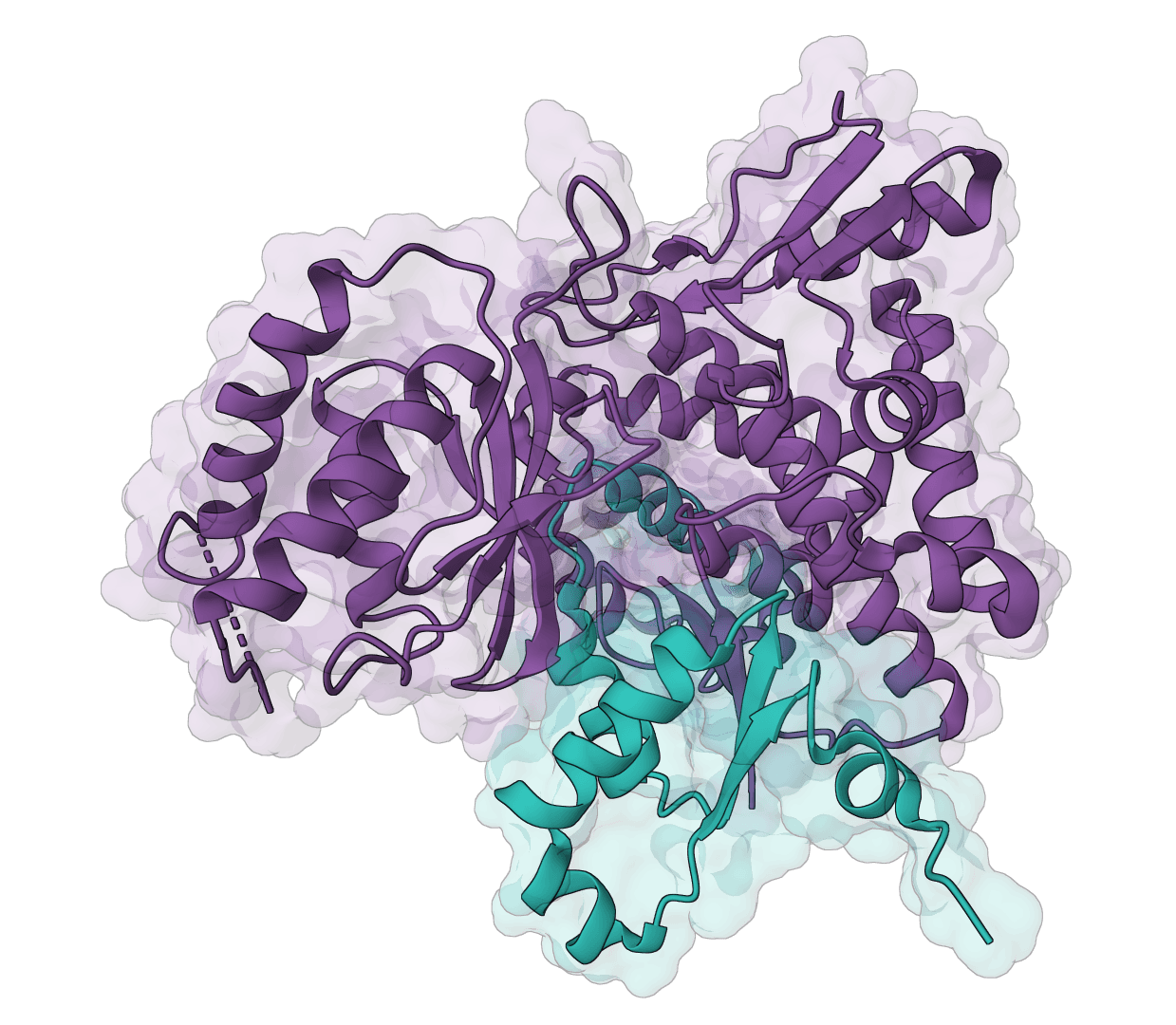
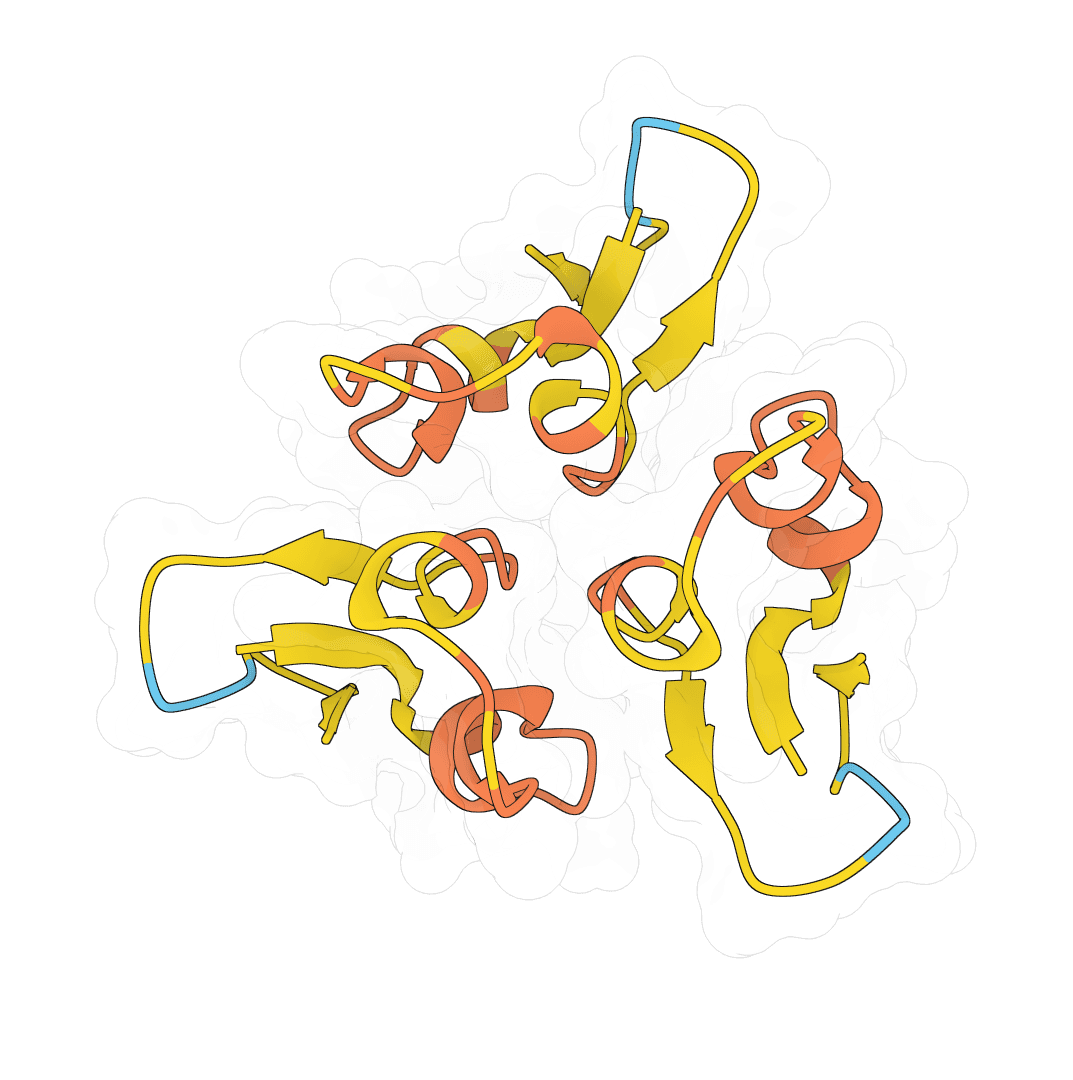
Open-source structure prediction neural network for proteins, nucleic acids, and small molecules. State-of-the-art accuracy with multi-chain support.
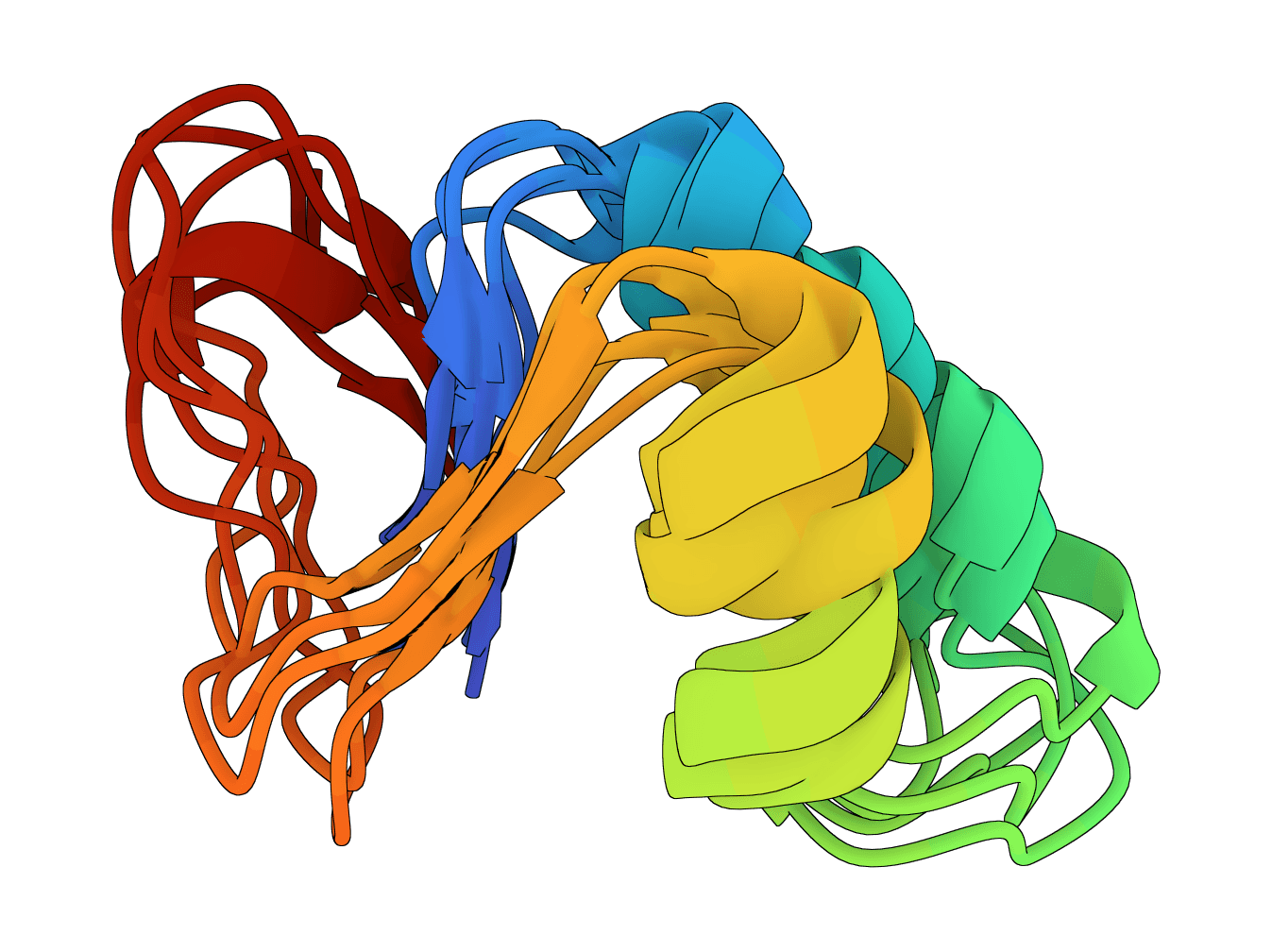
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
AlphaFold2 is the protein structure prediction model that revolutionized structural biology when DeepMind released it in 2021. By analyzing evolutionary relationships encoded in multiple sequence alignments (MSA), AlphaFold2 predicts 3D protein structures with accuracy rivaling experimental methods.
ProteinIQ runs ColabFold with two access paths. Guest and free-plan runs default to single-sequence inference with no database search. Paid plans can generate MSAs through MMseqs2, including the broad UniRef+Environmental mode used for the strongest evolutionary context.
AlphaFold2 supports both single-chain (monomer) and multi-chain (multimer) predictions. For complex structures involving ligands, DNA, or RNA, consider newer models like Boltz-2 or Chai-1 which extend beyond protein-only predictions.
Provide one or more protein sequences to generate ranked AlphaFold2 monomer or multimer structures online. Free runs use single-sequence mode, while paid plans can add MMseqs2 database searches for MSA-assisted prediction. Results include PDB structures, pLDDT, PAE, pTM, and ipTM metrics where applicable.
AlphaFold2 extracts coevolutionary information from MSAs—when two residue positions consistently change together across evolution, they're likely in spatial contact. The model transforms this evolutionary signal into accurate 3D coordinates.
The core innovation is the Evoformer module, a neural network that processes both sequence alignments and pairwise residue relationships simultaneously. Through 48 repeated blocks, information flows between these two representations, building increasingly refined structural features.
The MSA representation captures which residues are evolutionarily constrained and how they co-vary. The pair representation accumulates spatial relationship predictions between every residue pair.
After Evoformer processing, the structure module converts learned features into 3D atomic coordinates. It predicts backbone frames (position and orientation for each residue) and refines side-chain positions.
The prediction process includes recycling—feeding outputs back through the network for iterative refinement. Each recycle step improves the structure, particularly for challenging regions where initial predictions may be uncertain.
AlphaFold2-Multimer extends the base model to protein complexes. It uses paired MSAs that capture inter-chain coevolution, helping the model predict how chains interact. The model automatically detects if your input contains multiple chains and switches to multimer mode.
Provide protein sequences via text input (raw sequence or FASTA format), file upload (.fasta, .fa, .txt, .a3m), or PDB ID fetch from RCSB.
For multimer predictions, add multiple chains as separate inputs. Chain IDs are assigned automatically (A, B, C...). You can predict up to 10 chains in a single job.
3; increase to 6–12 for challenging predictions.Auto selects the appropriate variant based on your input—ptm for single chains, multimer v3 for complexes. Force a specific type only if you have a reason (e.g., benchmarking).MSA quality is the single most important factor for prediction accuracy. Deeper alignments with more homologous sequences generally produce better predictions.
No MSA disables sequence search and makes AlphaFold2 behave like a single-sequence model, similar to ESMFold.UniRef+Environmental searches the broadest set, including metagenomic sequences, and is the paid default. UniRef only is faster but may miss distant homologs. Remote MSA modes use twice the base credits shown for single-sequence mode.Unpaired+Paired combines chain-specific and cross-chain coevolution signals. Paired only uses only sequences where both chains appear together. Unpaired only treats chains independently.AlphaFold2 generates multiple ranked predictions, each with confidence metrics that help you assess reliability.
The predicted Local Distance Difference Test (pLDDT) estimates how accurate each residue's position is, stored in the B-factor column of output PDB files.
| pLDDT | Confidence | Interpretation |
|---|---|---|
| > 90 | Very high | High confidence in backbone and side chains |
| 70–90 | Good | Backbone likely correct; side chains may vary |
| 50–70 | Low | Structural uncertainty; interpret with caution |
| < 50 | Very low | Often indicates disorder or flexibility |
Residues with pLDDT below 50 frequently represent intrinsically disordered regions (IDRs) that lack fixed structure rather than prediction failures.
The predicted Template Modeling score (pTM) estimates overall structural accuracy, ranging from 0 to 1. Values above 0.7 typically indicate reliable predictions.
For multimers, the interface pTM (ipTM) specifically assesses inter-chain interface quality. The combined ranking score weights both metrics: ranking_score = 0.8 × ipTM + 0.2 × pTM. Combined scores above 0.62 suggest reliable complex predictions.
The Predicted Aligned Error matrix shows expected position errors between all residue pairs. Low PAE values (blue in visualizations) indicate confident relative positioning; high values (red) suggest uncertainty.
PAE is particularly useful for:
AlphaFold2 ranks predictions by confidence scores. We recommend examining the top 2–3 structures rather than only the #1 result—especially for flexible proteins or novel folds where ranking may be uncertain.
| Feature | AlphaFold2 | ESMFold | Boltz-2 | Chai-1 |
|---|---|---|---|---|
| Speed | Fast without MSA; moderate with MSA | Very fast (~seconds) | Moderate | Moderate |
| MSA required | Optional; paid modes add database search | No | Optional | Optional |
| Accuracy | Depends on target and MSA mode | Good (~0.95 TM) | Excellent | Excellent |
| Multimer support | Yes | Yes | Yes | Yes |
| Ligand prediction | No | No | Yes | Yes |
| DNA/RNA support | No | No | Yes | Yes |
Use AlphaFold2 when the established protein-only prediction workflow is needed. MSA-assisted paid modes are especially valuable for natural proteins with detectable evolutionary homologs; free single-sequence mode is faster but may be less accurate on difficult targets.
Use ESMFold when speed matters most or you're working with designed proteins that lack evolutionary relatives. ESMFold skips MSA generation entirely.
Use MiniFold when a fast single-sequence monomer predictor is enough and AlphaFold2's MSA search would be unnecessary overhead.
Use Boltz-2 or Chai-1 when you need to predict complexes involving small molecules, DNA, or RNA, or when you want binding affinity estimates alongside structure predictions.
Use OpenFold 3 or Protenix when you want open-source implementations of AlphaFold3-like capabilities for multi-molecule complexes.
AlphaFold2 requires evolutionarily related sequences for optimal performance. Predictions for de novo designed proteins or sequences without detectable homologs will have lower confidence.
The model predicts static structures and may not capture conformational changes, allosteric states, or dynamics. Regions with pLDDT below 50 often represent genuine flexibility rather than prediction errors.
AlphaFold2 cannot predict ligand binding, post-translational modifications, or the effects of small molecules on structure. For these applications, use newer models like Boltz-2.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
Open-source structure prediction neural network for proteins, nucleic acids, and small molecules. State-of-the-art accuracy with multi-chain support.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.