Protenix v2 is ByteDance Research's enhanced open-source biomolecular structure prediction model, released under the Apache 2.0 license as part of the Protenix project. It predicts 3D structures of complexes containing proteins, DNA, RNA, small molecule ligands, and metal ions from sequence or structural inputs, all in a single run.
AlphaFold 3 itself has restricted access and prohibits commercial use. Protenix follows the same general all-atom structure-prediction direction with open-source code and Apache-licensed model parameters, making it a practical option for open-ended research and applications where AlphaFold 3's terms would be limiting. ProteinIQ now runs Protenix v2 by default while keeping v1, mini, and tiny checkpoints available for comparison or faster exploratory runs.
ProteinIQ runs Protenix v2 on GPU infrastructure with no local installation. Add one or more molecules, optionally configure seeds and model, then submit. Results arrive as downloadable CIF files ranked by confidence score, viewable directly in the 3D structure viewer. Each ranked prediction also includes the Protenix summary-confidence JSON, and you can optionally export atom-level confidence JSON for deeper inspection. MSA search is off by default; enabling it can add several minutes but may improve protein predictions that benefit from evolutionary context.
Each molecule is added as a separate entity. Up to 10 entries of each type are supported.
| Input | Accepted formats |
|---|---|
| Protein | FASTA sequence, PDB file, or 4-letter RCSB PDB ID |
| Ligand | SMILES string, CCD code (e.g. ATP), SDF/MOL file, or PubChem compound ID |
| DNA | FASTA sequence (A, T, G, C) |
| RNA | FASTA sequence (A, U, G, C) |
| Ion | CCD code (e.g. ZN, MG, CA, FE) |
| Protein paired MSA | Optional uploaded or pasted .a3m alignment attached to a protein chain label |
| Protein unpaired MSA | Optional uploaded or pasted .a3m alignment attached to a protein chain label |
| RNA MSA | Optional uploaded or pasted .a3m alignment attached to an RNA chain label |
For ligand files, Protenix requires a 3D conformer. 2D SDF files (flat structure with no z-coordinates) will fail at featurization.
Precomputed alignments attach to the chain letters shown on the main protein or RNA molecule cards. If there is only one eligible protein or RNA chain, you can leave the MSA target label blank and ProteinIQ will attach it automatically. Protein paired MSA and protein unpaired MSA are separate native inputs; RNA only supports unpaired RNA-MSA in this pass. Template files are still out of scope here.
| Setting | Description |
|---|---|
Model seeds | Number of random seeds (1-5, default 1). Each seed independently samples the diffusion trajectory and produces a distinct set of structures. |
Model | Model variant. Protenix v2 is the default. See model options below. |
Use MSA | Whether to run multiple sequence alignment for protein chains (default off). Adds 2-5 minutes but can improve accuracy, especially for proteins with many homologs. |
Samples per seed | Structures generated per seed (1-5, default 5). Higher values increase output diversity. |
Precision | BF16 (default, faster) or FP32 (full precision). Negligible accuracy difference on A10G hardware. |
Training-Free Guidance | Applies physical constraints (steric, torsion, distance) during diffusion sampling to improve geometric plausibility. Off by default; adds compute overhead. |
Export atom confidence JSON | Saves the full_data JSON for each prediction. Useful for debugging or downstream analysis, but larger than the default summary metrics. |
| Model | Parameters | Training cutoff | When to use |
|---|---|---|---|
| Protenix v2 | 464M | 2021-09-30 | Default. Highest-accuracy model for most targets, with stronger antibody-antigen and ligand plausibility performance reported by ByteDance. |
| Protenix v1 base | 368M | 2021-09-30 | Baseline v1 model for comparison and lower compute cost. |
| Protenix v1 base (2025 data) | 368M | 2025-06-30 | Targets with templates deposited after 2021. |
| Protenix mini | 135M | 2021-09-30 | Rapid screening, large sequences, or quick estimates before a full run. Roughly 40% faster. |
Protenix v2 is the default model on ProteinIQ. It is a 464M-parameter model released in April 2026, with the same 2021-09-30 training cutoff used for fair comparison to earlier Protenix models.
The Protenix documentation reports stronger antibody-antigen performance and improved ligand plausibility compared with Protenix v1, while retaining MSA, RNA MSA, and template support. It also works with the Training-Free Guidance features introduced in the Protenix 2.0.0 package.
Protenix v1 remains available as a lower-compute comparison point. Its public checkpoints include the base 368M models, the 2025 cutoff model, and smaller mini/tiny variants.
Protenix implements the AlphaFold 3 architecture, which replaces the iterative refinement of AlphaFold 2 with a diffusion-based structure generation process. Prediction starts from randomly initialized atom coordinates and progressively denoises them into a coherent structure over multiple cycles.
For protein inputs, Protenix queries external MSA databases to find evolutionarily related sequences. Conserved residues and co-evolving residue pairs reveal spatial constraints that guide the structure prediction. The MSA step connects to an external server and is the primary source of runtime variability, typically adding 2-5 minutes per prediction.
If you already have alignments, ProteinIQ can use precomputed .a3m files instead of forcing a new search. Protein chains can receive separate paired and unpaired MSAs, while RNA chains can receive an unpaired RNA-MSA. These uploads are written to temporary files and referenced through the pairedMsaPath and unpairedMsaPath JSON fields used by Protenix.
Protenix outputs per-atom and per-residue confidence estimates derived from the same network that produces the structure. These are not validated against experimental data for each prediction; they reflect the model's internal consistency. A high-confidence prediction that matches an incorrect prior (e.g. a homolog with a different binding mode) may score well while being physically wrong.
Protenix outputs ranked CIF files together with the native summary-confidence JSON for each prediction.
| Metric | Scope | Interpretation |
|---|---|---|
| pLDDT | Per-residue, 0-100 | >90: high confidence. 70-90: backbone likely correct. 50-70: low confidence. <50: likely disordered or unreliable. |
| pTM | Whole structure, 0-1 | >0.5 suggests the overall fold is correct. |
| ipTM | Inter-chain interfaces, 0-1 | >0.7 indicates a reliable interface prediction. Particularly relevant for protein-protein and protein-ligand complexes. |
ProteinIQ also preserves the additional native metrics and flags from summary_confidence, including ranking_score, gpde, per-chain confidence summaries, pairwise interface scores, clash flags, disorder estimates, and the recycle count used for the prediction.
When running multiple seeds, structural agreement across the top-ranked predictions is a better indicator of reliability than any single confidence score. Divergent predictions across seeds suggest the structure is genuinely uncertain.
These completed jobs show how Protenix v2 handles protein-protein, protein-ligand, and protein-RNA systems. Open any example to inspect all three ranked predictions and download the CIF and confidence files.
This example predicts the well-characterized complex between the 110-residue barnase ribonuclease and its 89-residue barstar inhibitor. It demonstrates a compact protein-protein interface where evolutionary context materially improves the prediction.
Use MSA = on to supply evolutionary context; Samples per seed = 3 to return a concise ranked set; Export atom confidence JSON = on for residue- and atom-level inspection
The top prediction has a ranking_score of 0.975, pLDDT of 96.0, pTM of 0.977, and ipTM of 0.975, with no clash flag. The three displayed ranking scores round to the same value, showing consistent sampling for this run. These scores indicate strong model confidence in the fold and interface, but they do not replace comparison with an experimental complex structure.
This example submits the HIV-1 protease sequence as two copies together with stereospecific darunavir SMILES. It is useful for seeing how a protein-ligand prediction can return a plausible protein fold while remaining uncertain about the interaction.
Samples per seed = 3 to compare alternative predictions; Export atom confidence JSON = on for detailed confidence analysis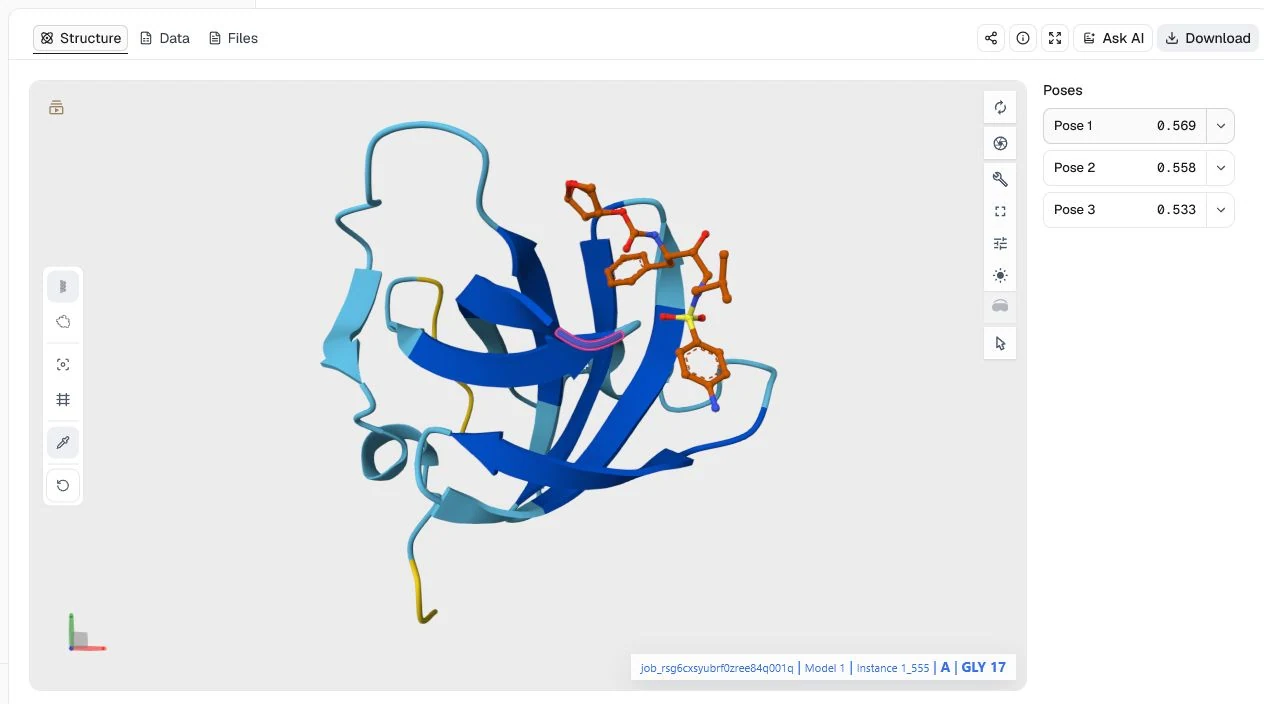
The top prediction has a ranking_score of 0.569, pLDDT of 80.0, pTM of 0.713, and ipTM of 0.533. The orange ligand is visible near the protein surface, but the interface score is below the 0.7 confidence heuristic used above. Treat this as an exploratory binding hypothesis: the run does not establish the experimental pose, binding affinity, or antiviral activity.
This protein-RNA example combines two copies of the MS2 bacteriophage coat protein with its 19-nucleotide operator hairpin. It demonstrates joint prediction across polymer types without an MSA search.
ACAUGAGGAUCACCCAUGU operator RNASamples per seed = 3 to compare the sampled complexes; Export atom confidence JSON = on for detailed confidence analysis
The RNA hairpin extends across the predicted protein complex in the Structure view. The top result has a ranking_score of 0.809, pLDDT of 81.2, pTM of 0.797, and ipTM of 0.812; the other two ranking scores are 0.804 and 0.802. The high interface score and agreement across the three samples support this model as a useful structural hypothesis, not experimental confirmation of the RNA-binding geometry.
The main differences are molecule scope, evolutionary inputs, guidance options, and whether the result describes one structure, an ensemble, or binding affinity.
| Tool | Best for | Key distinction |
|---|---|---|
| Protenix v2 | General all-atom complexes | Multiple model sizes, protein and RNA MSAs, optional atom confidence; no affinity prediction or user restraints. |
| Boltz-2 | Protein-ligand structure and affinity | Returns binder probability and affinity estimates; supports templates, constraints, and CCD covalent bonds. |
| Chai-1 | Guided complex prediction | Combines MSA and template search with contact, pocket, and modified-residue inputs. |
| OpenFold 3 | Open AlphaFold 3 research workflows | Trainable research preview with automatic MSA generation and low-memory inference. |
| RosettaFold3 | Alternative all-atom complex prediction | Handles proteins, DNA, RNA, and ligands with multi-model sampling. |
| IntelliFold 2 | Fast multi-component prediction | Offers a fast Flash model, optional MSA generation, and ranked complex structures. |
| AlphaFold2 | Protein monomers and multimers | Established protein-only workflow with single-sequence or MSA-assisted prediction. |
| ESMFold2 | Protein chains and protein complexes | Language-model folding with no MSA search and optional PAE and pair-chain ipTM outputs. |
| ESMFold | Rapid single-sequence protein folding | Predicts protein structures without an MSA and returns PDB, pLDDT, and optional PAE. |
| AlphaFlow | Protein conformational ensembles | Samples multiple structures to represent flexibility rather than one static prediction. |
| HighFold | Cyclic peptide structures | Models head-to-tail cyclization and disulfide constraints with an AlphaFold2-derived method. |
Protenix v2 is a strong default for unconstrained complexes containing several molecule types. Boltz-2 is the clearer choice for ligand affinity or pocket guidance, while Chai-1 is useful when templates, known contacts, or modified residues should influence the model.
Folding models predict structures from sequences and chemical inputs. For docking into a prepared receptor, AutoDock Vina searches a defined box and reports scores in kcal/mol, while DiffDock predicts ligand poses without a predefined box.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
OpenFold-3 is an open-source AI model for biomolecular structure prediction, aiming to reproduce AlphaFold3. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Open-source structure prediction neural network for proteins, nucleic acids, and small molecules. State-of-the-art accuracy with multi-chain support.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
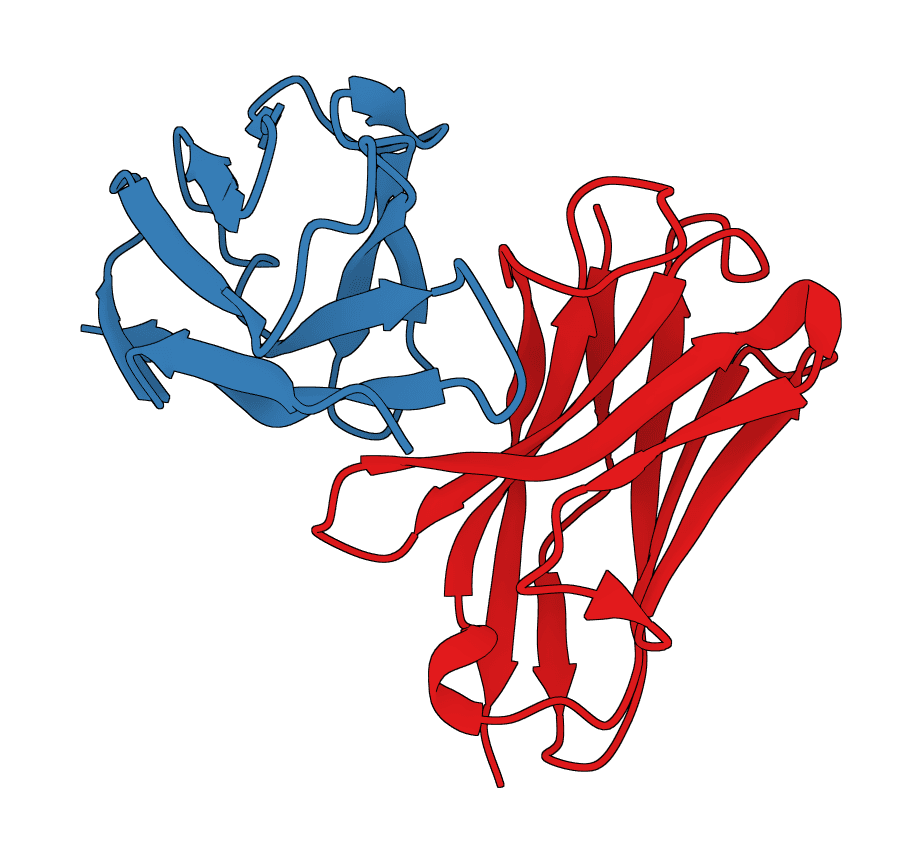
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
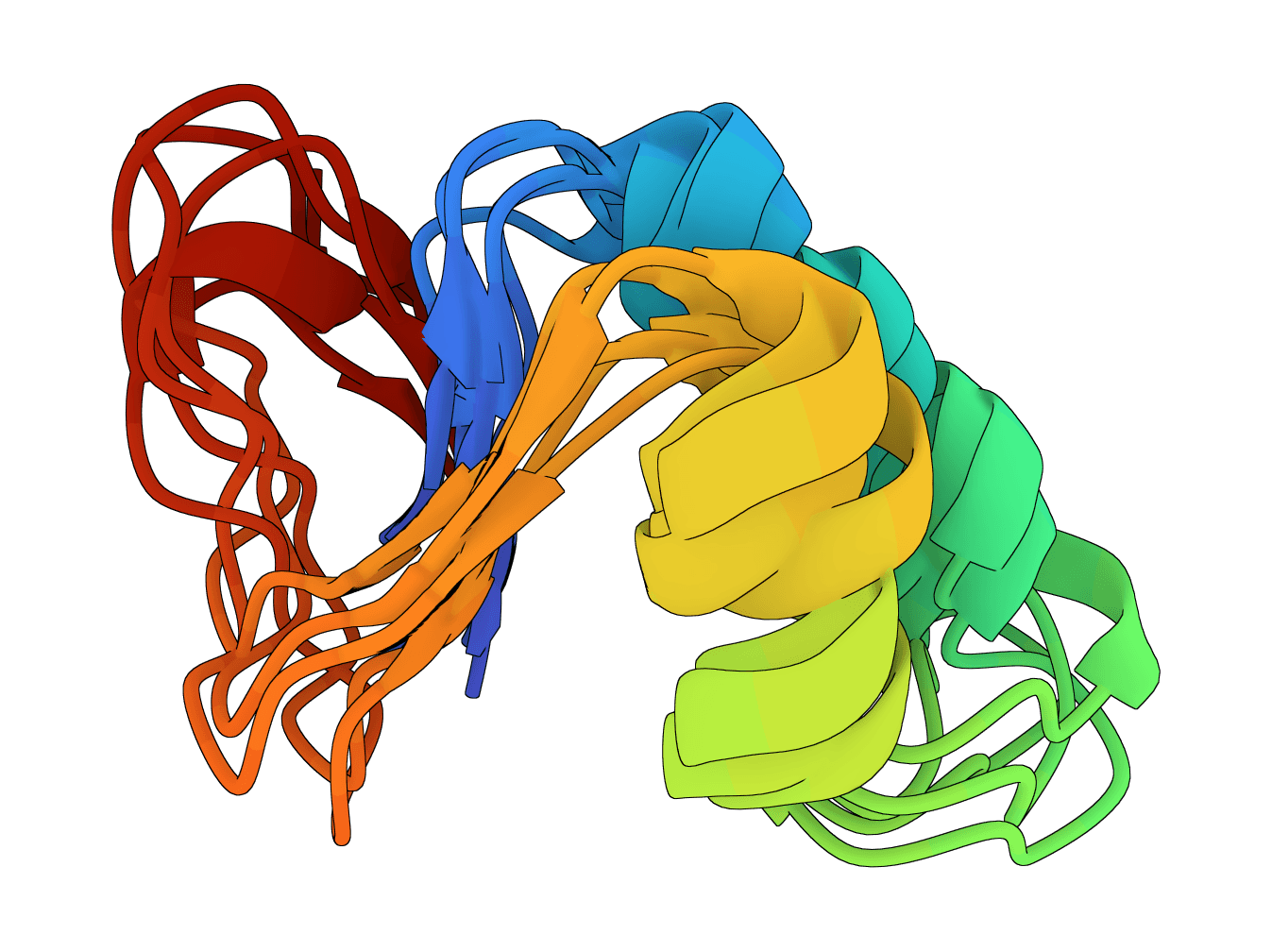
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
Protenix v2 is ByteDance Research's enhanced open-source biomolecular structure prediction model, released under the Apache 2.0 license as part of the Protenix project. It predicts 3D structures of complexes containing proteins, DNA, RNA, small molecule ligands, and metal ions from sequence or structural inputs, all in a single run.
AlphaFold 3 itself has restricted access and prohibits commercial use. Protenix follows the same general all-atom structure-prediction direction with open-source code and Apache-licensed model parameters, making it a practical option for open-ended research and applications where AlphaFold 3's terms would be limiting. ProteinIQ now runs Protenix v2 by default while keeping v1, mini, and tiny checkpoints available for comparison or faster exploratory runs.
ProteinIQ runs Protenix v2 on GPU infrastructure with no local installation. Add one or more molecules, optionally configure seeds and model, then submit. Results arrive as downloadable CIF files ranked by confidence score, viewable directly in the 3D structure viewer. Each ranked prediction also includes the Protenix summary-confidence JSON, and you can optionally export atom-level confidence JSON for deeper inspection. MSA search is off by default; enabling it can add several minutes but may improve protein predictions that benefit from evolutionary context.
Each molecule is added as a separate entity. Up to 10 entries of each type are supported.
| Input | Accepted formats |
|---|---|
| Protein | FASTA sequence, PDB file, or 4-letter RCSB PDB ID |
| Ligand | SMILES string, CCD code (e.g. ATP), SDF/MOL file, or PubChem compound ID |
| DNA | FASTA sequence (A, T, G, C) |
| RNA | FASTA sequence (A, U, G, C) |
| Ion | CCD code (e.g. ZN, MG, CA, FE) |
| Protein paired MSA | Optional uploaded or pasted .a3m alignment attached to a protein chain label |
| Protein unpaired MSA | Optional uploaded or pasted .a3m alignment attached to a protein chain label |
| RNA MSA | Optional uploaded or pasted .a3m alignment attached to an RNA chain label |
For ligand files, Protenix requires a 3D conformer. 2D SDF files (flat structure with no z-coordinates) will fail at featurization.
Precomputed alignments attach to the chain letters shown on the main protein or RNA molecule cards. If there is only one eligible protein or RNA chain, you can leave the MSA target label blank and ProteinIQ will attach it automatically. Protein paired MSA and protein unpaired MSA are separate native inputs; RNA only supports unpaired RNA-MSA in this pass. Template files are still out of scope here.
| Setting | Description |
|---|---|
Model seeds | Number of random seeds (1-5, default 1). Each seed independently samples the diffusion trajectory and produces a distinct set of structures. |
Model | Model variant. Protenix v2 is the default. See model options below. |
Use MSA | Whether to run multiple sequence alignment for protein chains (default off). Adds 2-5 minutes but can improve accuracy, especially for proteins with many homologs. |
Samples per seed | Structures generated per seed (1-5, default 5). Higher values increase output diversity. |
Precision | BF16 (default, faster) or FP32 (full precision). Negligible accuracy difference on A10G hardware. |
Training-Free Guidance | Applies physical constraints (steric, torsion, distance) during diffusion sampling to improve geometric plausibility. Off by default; adds compute overhead. |
Export atom confidence JSON | Saves the full_data JSON for each prediction. Useful for debugging or downstream analysis, but larger than the default summary metrics. |
| Model | Parameters | Training cutoff | When to use |
|---|---|---|---|
| Protenix v2 | 464M | 2021-09-30 | Default. Highest-accuracy model for most targets, with stronger antibody-antigen and ligand plausibility performance reported by ByteDance. |
| Protenix v1 base | 368M | 2021-09-30 | Baseline v1 model for comparison and lower compute cost. |
| Protenix v1 base (2025 data) | 368M | 2025-06-30 | Targets with templates deposited after 2021. |
| Protenix mini | 135M | 2021-09-30 | Rapid screening, large sequences, or quick estimates before a full run. Roughly 40% faster. |
Protenix v2 is the default model on ProteinIQ. It is a 464M-parameter model released in April 2026, with the same 2021-09-30 training cutoff used for fair comparison to earlier Protenix models.
The Protenix documentation reports stronger antibody-antigen performance and improved ligand plausibility compared with Protenix v1, while retaining MSA, RNA MSA, and template support. It also works with the Training-Free Guidance features introduced in the Protenix 2.0.0 package.
Protenix v1 remains available as a lower-compute comparison point. Its public checkpoints include the base 368M models, the 2025 cutoff model, and smaller mini/tiny variants.
Protenix implements the AlphaFold 3 architecture, which replaces the iterative refinement of AlphaFold 2 with a diffusion-based structure generation process. Prediction starts from randomly initialized atom coordinates and progressively denoises them into a coherent structure over multiple cycles.
For protein inputs, Protenix queries external MSA databases to find evolutionarily related sequences. Conserved residues and co-evolving residue pairs reveal spatial constraints that guide the structure prediction. The MSA step connects to an external server and is the primary source of runtime variability, typically adding 2-5 minutes per prediction.
If you already have alignments, ProteinIQ can use precomputed .a3m files instead of forcing a new search. Protein chains can receive separate paired and unpaired MSAs, while RNA chains can receive an unpaired RNA-MSA. These uploads are written to temporary files and referenced through the pairedMsaPath and unpairedMsaPath JSON fields used by Protenix.
Protenix outputs per-atom and per-residue confidence estimates derived from the same network that produces the structure. These are not validated against experimental data for each prediction; they reflect the model's internal consistency. A high-confidence prediction that matches an incorrect prior (e.g. a homolog with a different binding mode) may score well while being physically wrong.
Protenix outputs ranked CIF files together with the native summary-confidence JSON for each prediction.
| Metric | Scope | Interpretation |
|---|---|---|
| pLDDT | Per-residue, 0-100 | >90: high confidence. 70-90: backbone likely correct. 50-70: low confidence. <50: likely disordered or unreliable. |
| pTM | Whole structure, 0-1 | >0.5 suggests the overall fold is correct. |
| ipTM | Inter-chain interfaces, 0-1 | >0.7 indicates a reliable interface prediction. Particularly relevant for protein-protein and protein-ligand complexes. |
ProteinIQ also preserves the additional native metrics and flags from summary_confidence, including ranking_score, gpde, per-chain confidence summaries, pairwise interface scores, clash flags, disorder estimates, and the recycle count used for the prediction.
When running multiple seeds, structural agreement across the top-ranked predictions is a better indicator of reliability than any single confidence score. Divergent predictions across seeds suggest the structure is genuinely uncertain.
These completed jobs show how Protenix v2 handles protein-protein, protein-ligand, and protein-RNA systems. Open any example to inspect all three ranked predictions and download the CIF and confidence files.
This example predicts the well-characterized complex between the 110-residue barnase ribonuclease and its 89-residue barstar inhibitor. It demonstrates a compact protein-protein interface where evolutionary context materially improves the prediction.
Use MSA = on to supply evolutionary context; Samples per seed = 3 to return a concise ranked set; Export atom confidence JSON = on for residue- and atom-level inspectionThe top prediction has a ranking_score of 0.975, pLDDT of 96.0, pTM of 0.977, and ipTM of 0.975, with no clash flag. The three displayed ranking scores round to the same value, showing consistent sampling for this run. These scores indicate strong model confidence in the fold and interface, but they do not replace comparison with an experimental complex structure.
This example submits the HIV-1 protease sequence as two copies together with stereospecific darunavir SMILES. It is useful for seeing how a protein-ligand prediction can return a plausible protein fold while remaining uncertain about the interaction.
Samples per seed = 3 to compare alternative predictions; Export atom confidence JSON = on for detailed confidence analysisThe top prediction has a ranking_score of 0.569, pLDDT of 80.0, pTM of 0.713, and ipTM of 0.533. The orange ligand is visible near the protein surface, but the interface score is below the 0.7 confidence heuristic used above. Treat this as an exploratory binding hypothesis: the run does not establish the experimental pose, binding affinity, or antiviral activity.
This protein-RNA example combines two copies of the MS2 bacteriophage coat protein with its 19-nucleotide operator hairpin. It demonstrates joint prediction across polymer types without an MSA search.
ACAUGAGGAUCACCCAUGU operator RNASamples per seed = 3 to compare the sampled complexes; Export atom confidence JSON = on for detailed confidence analysisThe RNA hairpin extends across the predicted protein complex in the Structure view. The top result has a ranking_score of 0.809, pLDDT of 81.2, pTM of 0.797, and ipTM of 0.812; the other two ranking scores are 0.804 and 0.802. The high interface score and agreement across the three samples support this model as a useful structural hypothesis, not experimental confirmation of the RNA-binding geometry.
The main differences are molecule scope, evolutionary inputs, guidance options, and whether the result describes one structure, an ensemble, or binding affinity.
| Tool | Best for | Key distinction |
|---|---|---|
| Protenix v2 | General all-atom complexes | Multiple model sizes, protein and RNA MSAs, optional atom confidence; no affinity prediction or user restraints. |
| Boltz-2 | Protein-ligand structure and affinity | Returns binder probability and affinity estimates; supports templates, constraints, and CCD covalent bonds. |
| Chai-1 | Guided complex prediction | Combines MSA and template search with contact, pocket, and modified-residue inputs. |
| OpenFold 3 | Open AlphaFold 3 research workflows | Trainable research preview with automatic MSA generation and low-memory inference. |
| RosettaFold3 | Alternative all-atom complex prediction | Handles proteins, DNA, RNA, and ligands with multi-model sampling. |
| IntelliFold 2 | Fast multi-component prediction | Offers a fast Flash model, optional MSA generation, and ranked complex structures. |
| AlphaFold2 | Protein monomers and multimers | Established protein-only workflow with single-sequence or MSA-assisted prediction. |
| ESMFold2 | Protein chains and protein complexes | Language-model folding with no MSA search and optional PAE and pair-chain ipTM outputs. |
| ESMFold | Rapid single-sequence protein folding | Predicts protein structures without an MSA and returns PDB, pLDDT, and optional PAE. |
| AlphaFlow | Protein conformational ensembles | Samples multiple structures to represent flexibility rather than one static prediction. |
| HighFold | Cyclic peptide structures | Models head-to-tail cyclization and disulfide constraints with an AlphaFold2-derived method. |
Protenix v2 is a strong default for unconstrained complexes containing several molecule types. Boltz-2 is the clearer choice for ligand affinity or pocket guidance, while Chai-1 is useful when templates, known contacts, or modified residues should influence the model.
Folding models predict structures from sequences and chemical inputs. For docking into a prepared receptor, AutoDock Vina searches a defined box and reports scores in kcal/mol, while DiffDock predicts ligand poses without a predefined box.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
OpenFold-3 is an open-source AI model for biomolecular structure prediction, aiming to reproduce AlphaFold3. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Open-source structure prediction neural network for proteins, nucleic acids, and small molecules. State-of-the-art accuracy with multi-chain support.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.