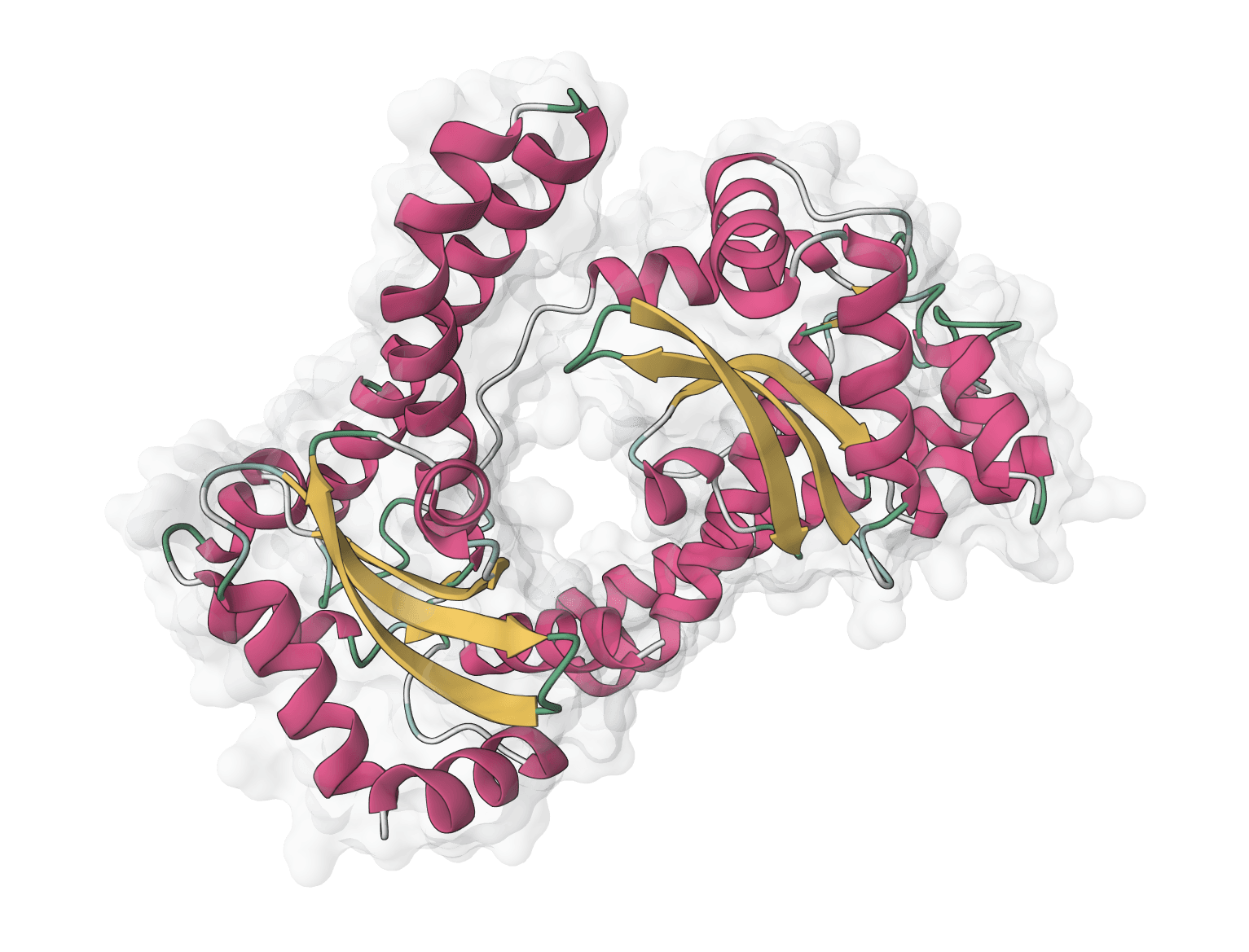
ESMFold is a protein structure prediction model developed by Meta AI Research (FAIR) and published in Science in 2023. The public esmfold_v1 release pairs a 3 billion-parameter ESM-2 language model stem with a folding trunk, allowing it to predict 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA).
The key advantage of ESMFold is speed. By eliminating the MSA generation step—which typically requires searching through large sequence databases—ESMFold achieves predictions up to 60x faster than AlphaFold2. This makes it particularly valuable for high-throughput screening and rapid prototyping.
ESMFold also supports multimer prediction, allowing you to predict structures of protein complexes with multiple chains. This is useful for studying protein-protein interactions and multi-subunit assemblies.
ESMFold takes a fundamentally different approach from MSA-based methods like AlphaFold2. Instead of explicitly searching for evolutionary homologs, it learns evolutionary patterns implicitly through language model pre-training.
The model is built on ESM-2, a transformer-based protein language model trained using masked residue prediction. During training, random amino acids in protein sequences are hidden, and the model learns to predict them from surrounding context—similar to how GPT-style models learn language patterns.
Through this training on millions of protein sequences, ESM-2 develops rich internal representations that capture evolutionary constraints and structural information. Remarkably, the attention patterns in these representations correspond to residue-residue contact maps in 3D structures.
ESMFold converts these learned representations into 3D coordinates through a structure module similar to AlphaFold2's. The model projects attention patterns onto known residue-residue contact maps derived from experimental structures, learning to translate sequence embeddings into atomic coordinates.
The prediction process includes a recycling phase where the model feeds its output back through the network multiple times. Each recycling step refines the predicted structure, allowing the model to resolve ambiguities in difficult regions.
On benchmark datasets, ESMFold achieves median TM-scores of 0.95 (vs 0.96 for AlphaFold2) and median RMSD of 1.74Å (vs 1.30Å for AlphaFold2). While slightly less accurate overall, ESMFold excels for proteins with limited sequence homology—precisely the cases where MSA generation provides little benefit anyway.
For high-confidence predictions (pLDDT > 70), ESMFold approaches experimental-level accuracy with backbone RMSD of 1.33Å.
You can provide protein sequences in several ways:
.fasta, .fa, .txt)For multimer prediction, add multiple chains as separate inputs. The tool passes them to ESMFold using the native multimer format, so chain breaks and chain IDs are preserved directly in the generated structure. The tool supports up to 10 chains per prediction.
Ambiguous or uncommon amino acids are preserved by mapping them to X before inference rather than silently deleting them, which keeps residue counts and numbering aligned with the input.
4 matches what was used during training and works well for most proteins. Setting this to 0 runs a single forward pass without extra recycling.Auto is recommended for most cases. Lower values (64, 32) reduce memory usage but increase runtime—useful if you encounter memory errors with long sequences.ESMFold outputs pLDDT (predicted Local Distance Difference Test) scores for each residue, stored in the B-factor field of the PDB file. These scores range from 0-100 and indicate local prediction confidence.
| pLDDT | Confidence | Interpretation |
|---|---|---|
| > 90 | Very high | Backbone and sidechains accurate |
| 70-90 | Good | Reliable backbone prediction |
| 50-70 | Low | Treat with caution |
| < 50 | Very low | Likely disordered or unstructured |
Regions with pLDDT below 50 often appear as extended ribbons and may represent intrinsically disordered regions rather than prediction failures. These regions may only adopt defined structures when bound to interaction partners.
Each prediction produces a PDB file containing atomic coordinates for all modeled residues. The B-factor column contains per-residue pLDDT scores on the standard 0-100 scale, which most molecular visualization tools can display as a color gradient.
The output summary also reports pTM and the maximum predicted aligned error. If you enable Save PAE matrix, the job includes a JSON file containing the full residue-level predicted_aligned_error matrix.
| Feature | ESMFold | Boltz-2 | Chai-1 |
|---|---|---|---|
| Speed | Very fast (~seconds) | Moderate (~20 sec) | Moderate |
| MSA required | No | Optional | Optional |
| Accuracy (TM-score) | ~0.95 | ~0.96 | ~0.95 |
| Multimer support | Yes | Yes | Yes |
| Ligand prediction | No | Yes | Yes |
| DNA/RNA support | No | Yes | Yes |
| Affinity prediction | No | Yes | No |
Use ESMFold when you need rapid structure predictions, are working with designed proteins lacking natural homologs, or want to quickly screen many sequences before running more expensive predictions.
For related single-sequence folding workflows, compare MiniFold for faster monomer prediction and ESMFold2 for the newer ESMFold-family implementation.
Use Boltz-2 when you need the highest accuracy, want to predict protein-ligand complexes, or need binding affinity estimates. Boltz-2's MSA generation improves accuracy for natural proteins.
Use Chai-1 when you're predicting multi-component complexes involving DNA, RNA, or small molecules, and don't need affinity predictions.
ESMFold trades some accuracy for speed. For applications requiring the highest possible accuracy—such as drug design or detailed mechanistic studies—MSA-based methods like Boltz-2 or AlphaFold2 may be more appropriate.
The model works best for globular proteins with clear evolutionary signatures encoded in the sequence. Predictions for intrinsically disordered regions, membrane proteins, or highly repetitive sequences may be less reliable.
ESMFold also cannot predict ligand binding poses or non-protein molecules. For protein-small molecule or protein-nucleic acid complexes, use Boltz-2 or Chai-1 instead.
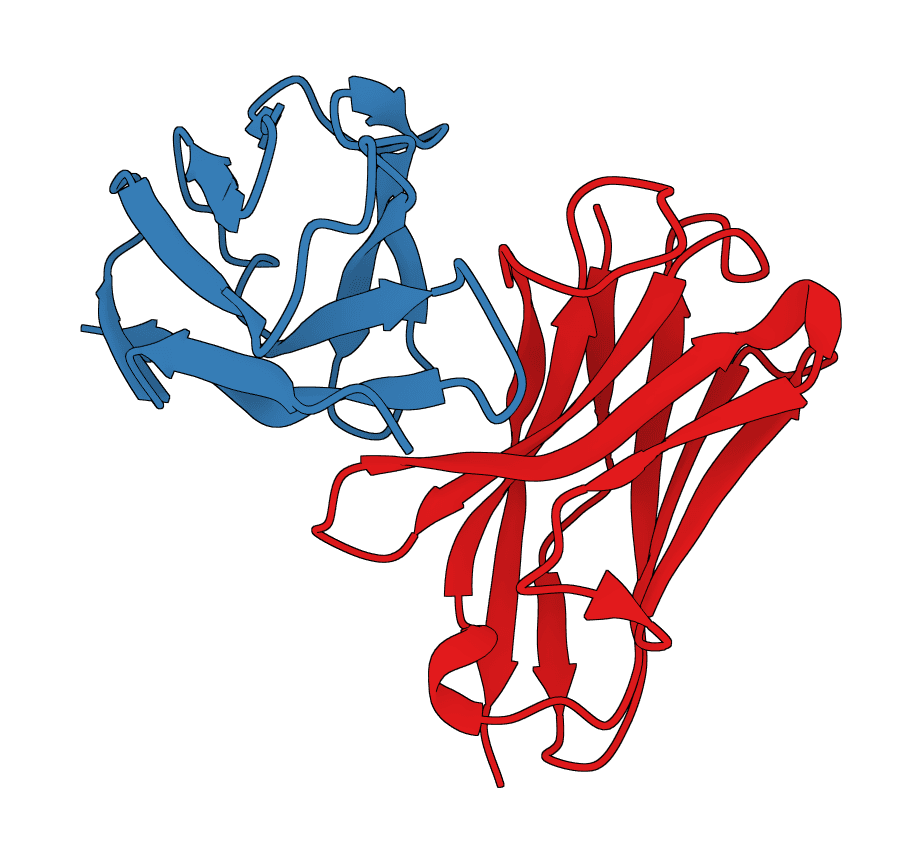
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
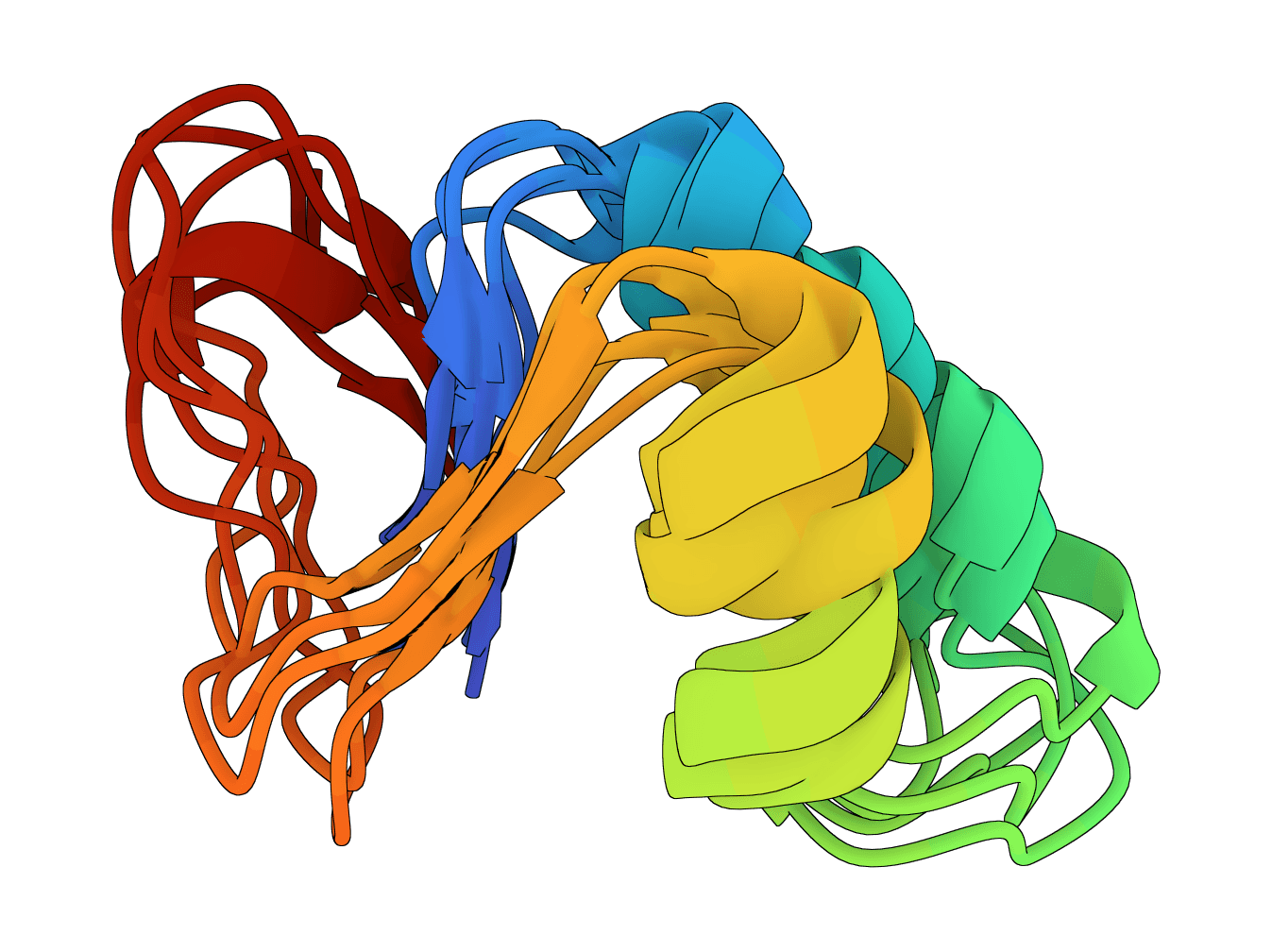
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
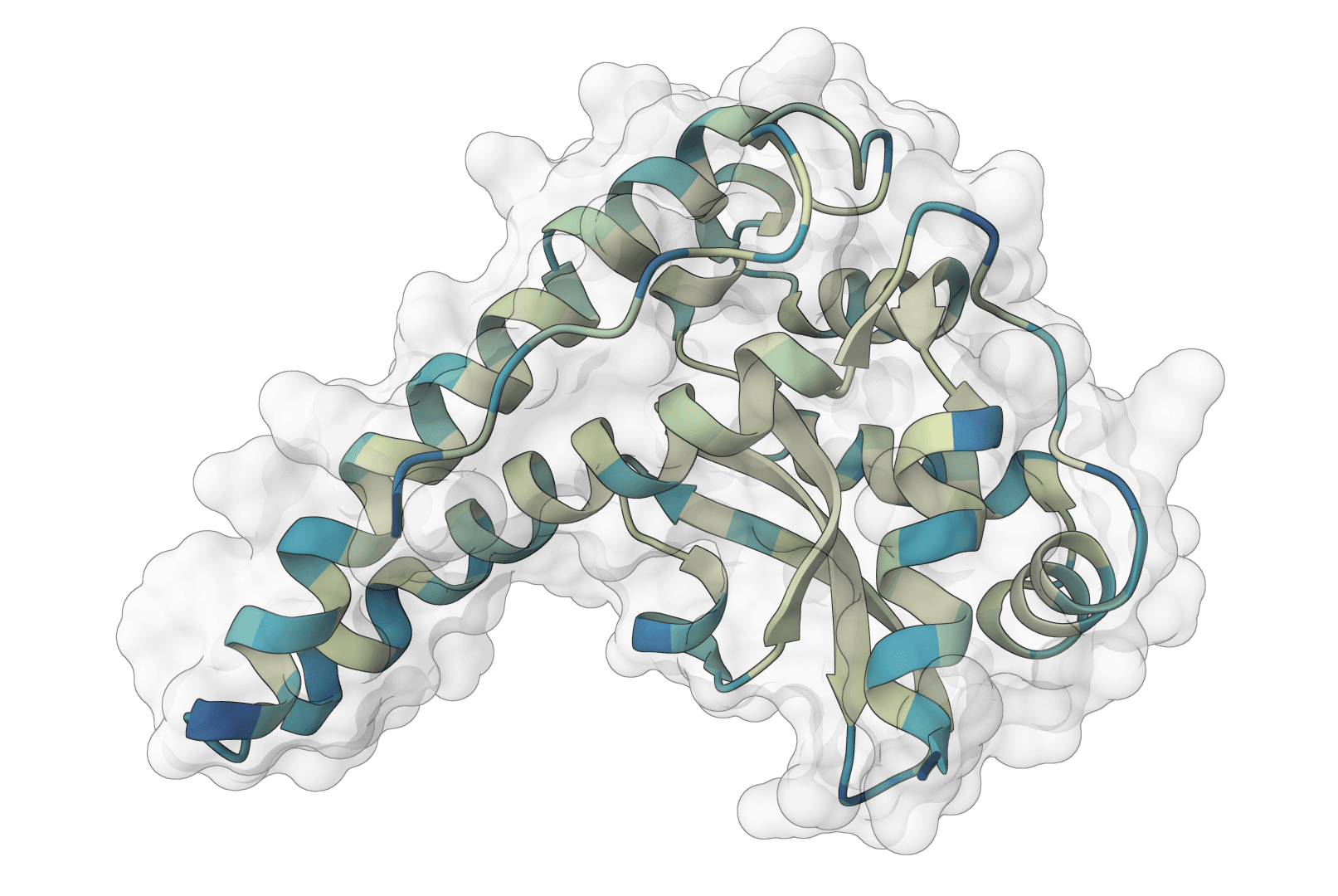
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
MiniFold is a fast single-sequence protein structure predictor that is 10-20x faster than ESMFold. It predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it ideal for rapid structure prediction.
ESMFold is a protein structure prediction model developed by Meta AI Research (FAIR) and published in Science in 2023. The public esmfold_v1 release pairs a 3 billion-parameter ESM-2 language model stem with a folding trunk, allowing it to predict 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA).
The key advantage of ESMFold is speed. By eliminating the MSA generation step—which typically requires searching through large sequence databases—ESMFold achieves predictions up to 60x faster than AlphaFold2. This makes it particularly valuable for high-throughput screening and rapid prototyping.
ESMFold also supports multimer prediction, allowing you to predict structures of protein complexes with multiple chains. This is useful for studying protein-protein interactions and multi-subunit assemblies.
ESMFold takes a fundamentally different approach from MSA-based methods like AlphaFold2. Instead of explicitly searching for evolutionary homologs, it learns evolutionary patterns implicitly through language model pre-training.
The model is built on ESM-2, a transformer-based protein language model trained using masked residue prediction. During training, random amino acids in protein sequences are hidden, and the model learns to predict them from surrounding context—similar to how GPT-style models learn language patterns.
Through this training on millions of protein sequences, ESM-2 develops rich internal representations that capture evolutionary constraints and structural information. Remarkably, the attention patterns in these representations correspond to residue-residue contact maps in 3D structures.
ESMFold converts these learned representations into 3D coordinates through a structure module similar to AlphaFold2's. The model projects attention patterns onto known residue-residue contact maps derived from experimental structures, learning to translate sequence embeddings into atomic coordinates.
The prediction process includes a recycling phase where the model feeds its output back through the network multiple times. Each recycling step refines the predicted structure, allowing the model to resolve ambiguities in difficult regions.
On benchmark datasets, ESMFold achieves median TM-scores of 0.95 (vs 0.96 for AlphaFold2) and median RMSD of 1.74Å (vs 1.30Å for AlphaFold2). While slightly less accurate overall, ESMFold excels for proteins with limited sequence homology—precisely the cases where MSA generation provides little benefit anyway.
For high-confidence predictions (pLDDT > 70), ESMFold approaches experimental-level accuracy with backbone RMSD of 1.33Å.
You can provide protein sequences in several ways:
.fasta, .fa, .txt)For multimer prediction, add multiple chains as separate inputs. The tool passes them to ESMFold using the native multimer format, so chain breaks and chain IDs are preserved directly in the generated structure. The tool supports up to 10 chains per prediction.
Ambiguous or uncommon amino acids are preserved by mapping them to X before inference rather than silently deleting them, which keeps residue counts and numbering aligned with the input.
4 matches what was used during training and works well for most proteins. Setting this to 0 runs a single forward pass without extra recycling.Auto is recommended for most cases. Lower values (64, 32) reduce memory usage but increase runtime—useful if you encounter memory errors with long sequences.ESMFold outputs pLDDT (predicted Local Distance Difference Test) scores for each residue, stored in the B-factor field of the PDB file. These scores range from 0-100 and indicate local prediction confidence.
| pLDDT | Confidence | Interpretation |
|---|---|---|
| > 90 | Very high | Backbone and sidechains accurate |
| 70-90 | Good | Reliable backbone prediction |
| 50-70 | Low | Treat with caution |
| < 50 | Very low | Likely disordered or unstructured |
Regions with pLDDT below 50 often appear as extended ribbons and may represent intrinsically disordered regions rather than prediction failures. These regions may only adopt defined structures when bound to interaction partners.
Each prediction produces a PDB file containing atomic coordinates for all modeled residues. The B-factor column contains per-residue pLDDT scores on the standard 0-100 scale, which most molecular visualization tools can display as a color gradient.
The output summary also reports pTM and the maximum predicted aligned error. If you enable Save PAE matrix, the job includes a JSON file containing the full residue-level predicted_aligned_error matrix.
| Feature | ESMFold | Boltz-2 | Chai-1 |
|---|---|---|---|
| Speed | Very fast (~seconds) | Moderate (~20 sec) | Moderate |
| MSA required | No | Optional | Optional |
| Accuracy (TM-score) | ~0.95 | ~0.96 | ~0.95 |
| Multimer support | Yes | Yes | Yes |
| Ligand prediction | No | Yes | Yes |
| DNA/RNA support | No | Yes | Yes |
| Affinity prediction | No | Yes | No |
Use ESMFold when you need rapid structure predictions, are working with designed proteins lacking natural homologs, or want to quickly screen many sequences before running more expensive predictions.
For related single-sequence folding workflows, compare MiniFold for faster monomer prediction and ESMFold2 for the newer ESMFold-family implementation.
Use Boltz-2 when you need the highest accuracy, want to predict protein-ligand complexes, or need binding affinity estimates. Boltz-2's MSA generation improves accuracy for natural proteins.
Use Chai-1 when you're predicting multi-component complexes involving DNA, RNA, or small molecules, and don't need affinity predictions.
ESMFold trades some accuracy for speed. For applications requiring the highest possible accuracy—such as drug design or detailed mechanistic studies—MSA-based methods like Boltz-2 or AlphaFold2 may be more appropriate.
The model works best for globular proteins with clear evolutionary signatures encoded in the sequence. Predictions for intrinsically disordered regions, membrane proteins, or highly repetitive sequences may be less reliable.
ESMFold also cannot predict ligand binding poses or non-protein molecules. For protein-small molecule or protein-nucleic acid complexes, use Boltz-2 or Chai-1 instead.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
MiniFold is a fast single-sequence protein structure predictor that is 10-20x faster than ESMFold. It predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it ideal for rapid structure prediction.