Analyze protein backbone phi/psi angles and classify favored, allowed, and outlier residues. Learn more
Run
Output
Configure inputs to begin
Set options on the left, then click “Generate”.
Analyze protein backbone phi/psi angles and classify favored, allowed, and outlier residues. Learn more
Configure inputs to begin
Set options on the left, then click “Generate”.
Analyze protein backbone phi/psi angles and classify favored, allowed, and outlier residues. Learn more
Configure inputs to begin
Set options on the left, then click “Generate”.
A Ramachandran plot is a two-dimensional visualization that maps the energetically allowed and forbidden backbone dihedral angles (φ and ψ) of the amino acid residues in a protein structure. Biochemists and structural biologists use it to study protein folding and to validate protein models. G.N. Ramachandran, C. Ramakrishnan, and V. Sasisekharan introduced it in 1963 in the Journal of Molecular Biology.
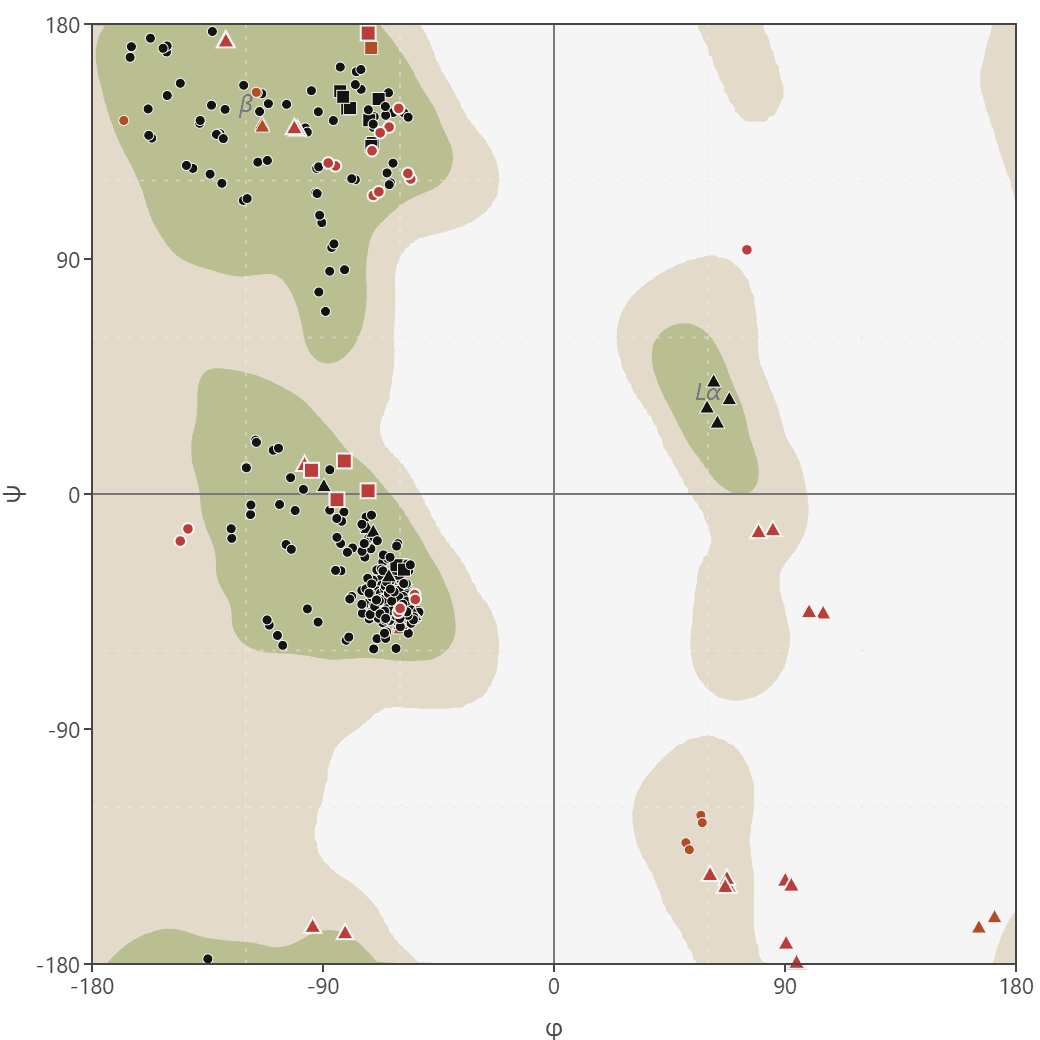
Most φ/ψ combinations force backbone atoms to clash, so they never occur in real structures. The combinations that remain form the allowed regions of the plot, which correspond to the common secondary structures: alpha helices, beta sheets, and turns.
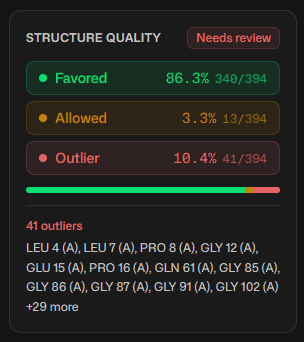
Ramachandran analysis is a standard validation step for structures from X-ray crystallography and cryo-EM. A well-refined high-resolution structure places more than 98% of its residues in favored regions. Residues that fall outside the allowed regions, called outliers, usually point to a modeling error or a genuine strained conformation that has a functional reason.
The protein backbone has three dihedral angles per residue. The phi (φ) angle is the rotation around the bond between the nitrogen and the α-carbon. The psi (ψ) angle is the rotation around the bond between the α-carbon and the carbonyl carbon. The omega (ω) angle at the peptide bond stays near 180° because of its partial double-bond character.
The φ angle is measured from four atoms: C(i-1), N(i), Cα(i), and C(i). The ψ angle uses N(i), Cα(i), C(i), and N(i+1). Because each angle depends on a neighboring residue, the first and last residue of every chain are missing one angle and do not appear on the plot.
Ramachandran's original analysis modeled atoms as hard spheres with van der Waals radii and sampled every φ/ψ combination. Many combinations are forbidden because the atoms would clash, a constraint called steric hindrance, leaving a small set of allowed ranges that map directly onto secondary structure. Each allowed region sits in a characteristic part of the plot.
| Conformation | Approximate φ | Approximate ψ | Plot location |
|---|---|---|---|
| Beta sheet | -120° | +120° | Top left |
| Right-handed alpha helix | -60° | -45° | Bottom left |
| Left-handed alpha helix | +60° | +45° | Top right |
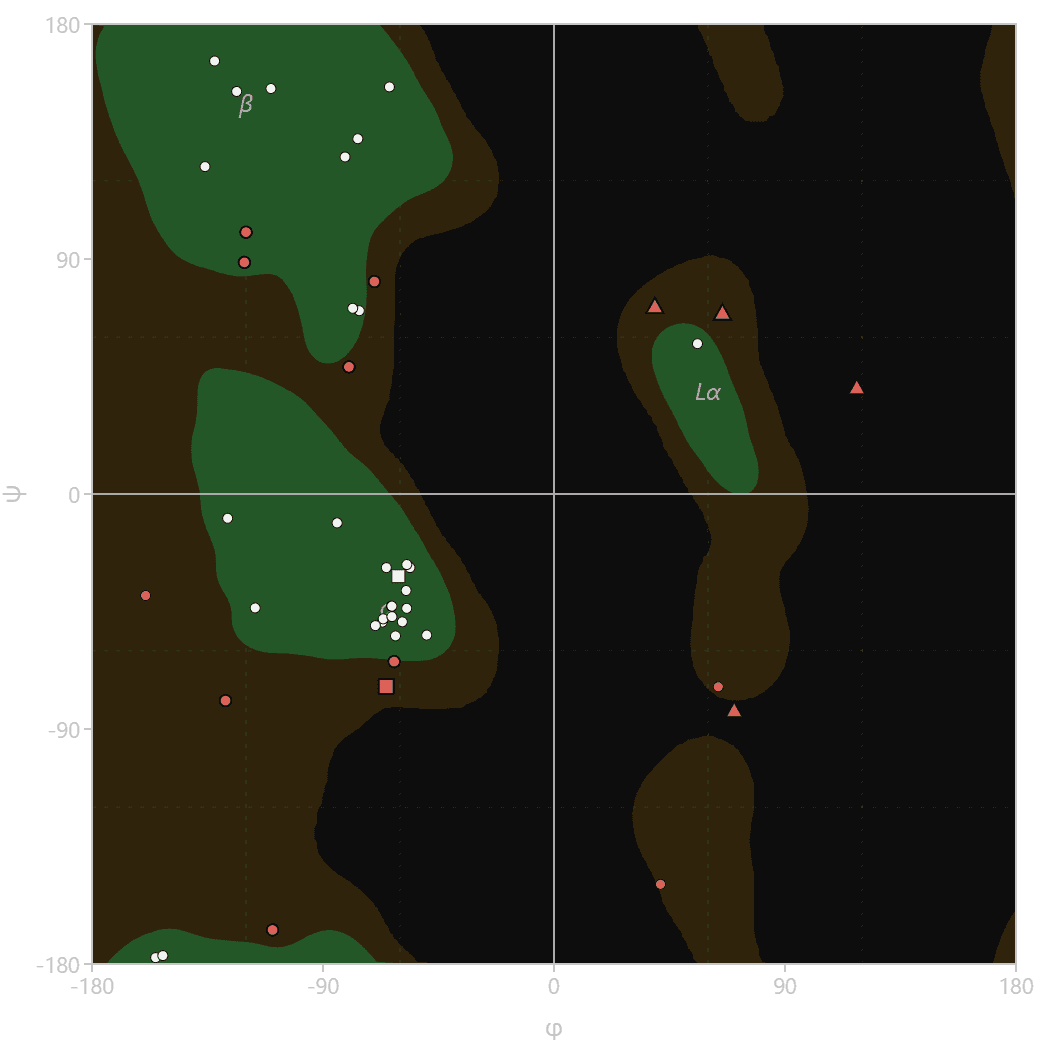
The allowed regions differ by residue type because of side-chain geometry. The tool classifies and plots each residue against the reference set for its own class.
| Class | Allowed region behavior |
|---|---|
| General | The standard distribution shared by most amino acids, with φ predominantly negative |
| Glycine | A single hydrogen side chain gives minimal steric hindrance, so the allowed region is large and nearly symmetric |
| Cis-proline | Proline following a cis peptide bond has its own compact reference distribution |
| Trans-proline | A cyclic side chain locks φ near -60°, confining it to a narrow strip |
| Pre-proline | Residues immediately before a proline shift due to contact with the proline ring |
| Isoleucine or valine | β-branched side chains produce a distinct distribution from the general class |
The tool uses the six residue classes and favored/allowed thresholds from the cctbx rama8000 validation tables. In each residue-class view, classification uses the same data-derived contours drawn on the plot.
| Region | Population in well-refined structures | Meaning |
|---|---|---|
| Favored | ~98% of residues | Optimal backbone geometry |
| Allowed | ~2% of residues | Sterically possible but less common |
| Outlier | Remaining residues | Possible modeling error or a real strained conformation |
The tool reads PDB and mmCIF files with atomic coordinates and needs the complete backbone atoms (N, Cα, C) to calculate angles. Residues are connected using peptide-bond geometry rather than requiring consecutive author residue numbers. Enter a PDB ID to fetch its mmCIF structure directly from the RCSB Protein Data Bank, or upload your own file. For multi-model files such as NMR ensembles, only the first model is analyzed.
The plot opens on all residues at once. Each point is one residue's φ/ψ pair and is colored by its own residue-class classification, with outliers in red. Because the six classes have different reference regions, the combined view does not show a potentially misleading single background. Glycine and proline appear as distinct marker shapes.
The view selector switches the plot to a single residue class and draws that class's favored and allowed regions. Hover any point to read its name, number, chain, exact angles, residue class, and region. Display options filter by chain, toggle the reference regions and annotations, hide outliers, and adjust point size.
The sidebar reports the percentage of residues in each region and a quality verdict.
| Favored residues | Interpretation |
|---|---|
| Above 98% | Expected for high-quality structures |
| 95% to 98% | Acceptable for most purposes |
| Below 95% | May indicate structural problems that need investigation |
Flagged outliers are listed in the sidebar, and the data table tab lists every residue with its exact angles for sorting or export. Not every outlier is an error. Functionally important residues sometimes adopt strained conformations for catalysis or binding, so compare outlier positions with functional annotations before treating them as mistakes.
| Application | Purpose |
|---|---|
| Model validation | Assess crystal structures, NMR ensembles, and homology models |
| Structure refinement | Find residues that need manual adjustment during model building |
| Comparative analysis | Compare conformational states or mutants of the same protein |
| Teaching | Show how backbone geometry relates to secondary structure |
Ramachandran plot generation is free and runs entirely in your browser. No credits required.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.

Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
PoseBusters validates generated or docked molecular poses with chemically and structurally grounded quality checks for molecular geometry, intermolecular interactions, and optional reference-pose agreement.
Plot net charge vs pH for protein sequences. Visualize how protein charge changes across pH 0-14 and identify the isoelectric point (pI) where the net charge crosses zero.
Generate Kyte-Doolittle hydropathy plots to visualize hydrophobic and hydrophilic regions along protein sequences. Identify transmembrane domains and surface-exposed regions.
Generate hydrophobicity plots using 24 different amino acid scales. Visualize hydrophobic and hydrophilic regions for protein analysis, epitope prediction, and membrane protein studies.
Generate amino acid property profiles using 42 different scales spanning hydrophobicity, secondary structure propensity, flexibility, polarity, surface accessibility, antigenicity, and more.
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.
A Ramachandran plot is a two-dimensional visualization that maps the energetically allowed and forbidden backbone dihedral angles (φ and ψ) of the amino acid residues in a protein structure. Biochemists and structural biologists use it to study protein folding and to validate protein models. G.N. Ramachandran, C. Ramakrishnan, and V. Sasisekharan introduced it in 1963 in the Journal of Molecular Biology.
Most φ/ψ combinations force backbone atoms to clash, so they never occur in real structures. The combinations that remain form the allowed regions of the plot, which correspond to the common secondary structures: alpha helices, beta sheets, and turns.
Ramachandran analysis is a standard validation step for structures from X-ray crystallography and cryo-EM. A well-refined high-resolution structure places more than 98% of its residues in favored regions. Residues that fall outside the allowed regions, called outliers, usually point to a modeling error or a genuine strained conformation that has a functional reason.
The protein backbone has three dihedral angles per residue. The phi (φ) angle is the rotation around the bond between the nitrogen and the α-carbon. The psi (ψ) angle is the rotation around the bond between the α-carbon and the carbonyl carbon. The omega (ω) angle at the peptide bond stays near 180° because of its partial double-bond character.
The φ angle is measured from four atoms: C(i-1), N(i), Cα(i), and C(i). The ψ angle uses N(i), Cα(i), C(i), and N(i+1). Because each angle depends on a neighboring residue, the first and last residue of every chain are missing one angle and do not appear on the plot.
Ramachandran's original analysis modeled atoms as hard spheres with van der Waals radii and sampled every φ/ψ combination. Many combinations are forbidden because the atoms would clash, a constraint called steric hindrance, leaving a small set of allowed ranges that map directly onto secondary structure. Each allowed region sits in a characteristic part of the plot.
| Conformation | Approximate φ | Approximate ψ | Plot location |
|---|---|---|---|
| Beta sheet | -120° | +120° | Top left |
| Right-handed alpha helix | -60° | -45° | Bottom left |
| Left-handed alpha helix | +60° | +45° | Top right |
The allowed regions differ by residue type because of side-chain geometry. The tool classifies and plots each residue against the reference set for its own class.
| Class | Allowed region behavior |
|---|---|
| General | The standard distribution shared by most amino acids, with φ predominantly negative |
| Glycine | A single hydrogen side chain gives minimal steric hindrance, so the allowed region is large and nearly symmetric |
| Cis-proline | Proline following a cis peptide bond has its own compact reference distribution |
| Trans-proline | A cyclic side chain locks φ near -60°, confining it to a narrow strip |
| Pre-proline | Residues immediately before a proline shift due to contact with the proline ring |
| Isoleucine or valine | β-branched side chains produce a distinct distribution from the general class |
The tool uses the six residue classes and favored/allowed thresholds from the cctbx rama8000 validation tables. In each residue-class view, classification uses the same data-derived contours drawn on the plot.
| Region | Population in well-refined structures | Meaning |
|---|---|---|
| Favored | ~98% of residues | Optimal backbone geometry |
| Allowed | ~2% of residues | Sterically possible but less common |
| Outlier | Remaining residues | Possible modeling error or a real strained conformation |
The tool reads PDB and mmCIF files with atomic coordinates and needs the complete backbone atoms (N, Cα, C) to calculate angles. Residues are connected using peptide-bond geometry rather than requiring consecutive author residue numbers. Enter a PDB ID to fetch its mmCIF structure directly from the RCSB Protein Data Bank, or upload your own file. For multi-model files such as NMR ensembles, only the first model is analyzed.
The plot opens on all residues at once. Each point is one residue's φ/ψ pair and is colored by its own residue-class classification, with outliers in red. Because the six classes have different reference regions, the combined view does not show a potentially misleading single background. Glycine and proline appear as distinct marker shapes.
The view selector switches the plot to a single residue class and draws that class's favored and allowed regions. Hover any point to read its name, number, chain, exact angles, residue class, and region. Display options filter by chain, toggle the reference regions and annotations, hide outliers, and adjust point size.
The sidebar reports the percentage of residues in each region and a quality verdict.
| Favored residues | Interpretation |
|---|---|
| Above 98% | Expected for high-quality structures |
| 95% to 98% | Acceptable for most purposes |
| Below 95% | May indicate structural problems that need investigation |
Flagged outliers are listed in the sidebar, and the data table tab lists every residue with its exact angles for sorting or export. Not every outlier is an error. Functionally important residues sometimes adopt strained conformations for catalysis or binding, so compare outlier positions with functional annotations before treating them as mistakes.
| Application | Purpose |
|---|---|
| Model validation | Assess crystal structures, NMR ensembles, and homology models |
| Structure refinement | Find residues that need manual adjustment during model building |
| Comparative analysis | Compare conformational states or mutants of the same protein |
| Teaching | Show how backbone geometry relates to secondary structure |
Ramachandran plot generation is free and runs entirely in your browser. No credits required.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
PoseBusters validates generated or docked molecular poses with chemically and structurally grounded quality checks for molecular geometry, intermolecular interactions, and optional reference-pose agreement.
Plot net charge vs pH for protein sequences. Visualize how protein charge changes across pH 0-14 and identify the isoelectric point (pI) where the net charge crosses zero.
Generate Kyte-Doolittle hydropathy plots to visualize hydrophobic and hydrophilic regions along protein sequences. Identify transmembrane domains and surface-exposed regions.
Generate hydrophobicity plots using 24 different amino acid scales. Visualize hydrophobic and hydrophilic regions for protein analysis, epitope prediction, and membrane protein studies.
Generate amino acid property profiles using 42 different scales spanning hydrophobicity, secondary structure propensity, flexibility, polarity, surface accessibility, antigenicity, and more.
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.