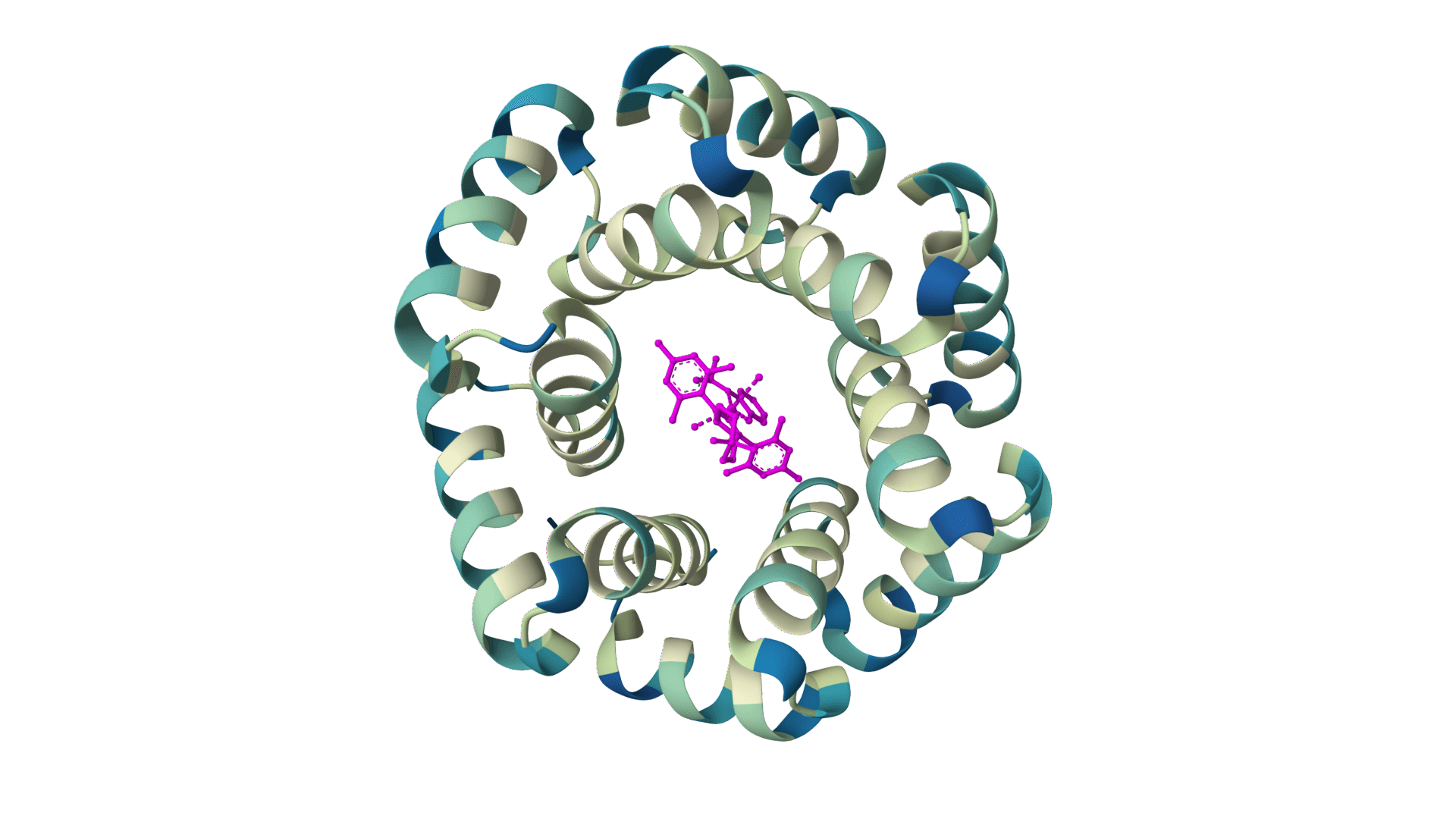
Design protein sequences around ligands, metals, and nucleotides for enzyme engineering and binding-site optimization. Learn more
Design protein sequences around ligands, metals, and nucleotides for enzyme engineering and binding-site optimization. Learn more
Design protein sequences around ligands, metals, and nucleotides for enzyme engineering and binding-site optimization. Learn more
Design thermostable protein sequences using ProteinMPNN trained on hyperthermophilic organism structures. Generates sequences optimized for improved thermal stability without requiring ligands or additional context.
Design protein sequences for given backbone structures using deep learning. Fast and accurate inverse folding with state-of-the-art sequence recovery (52.4%).
Specialized model for soluble protein sequence design. Trained exclusively on soluble proteins for optimized performance on cytoplasmic and extracellular proteins.
Design antibody heavy- and light-chain CDR sequences from an antibody-antigen complex with the IgDesign inverse-folding model.
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
Inverse folding with ESM-IF1. Design protein sequences for given 3D backbone structures using a geometric deep learning model. Generate multiple sequence variants optimized for your target structure.

ProFam-1 is a protein family language model for family-conditioned sequence generation. Provide a protein family in FASTA, A2M, or A3M format and generate new sequences with model likelihood scores for downstream ranking and screening.
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
All-atom generative diffusion model for protein design with complex constraints. Design binders, enzymes, and symmetric protein assemblies.
Design linear peptide binders for target proteins using a target sequence-conditioned masked language model. PepMLM generates peptide sequences optimized to bind specific protein targets based on ESM-2 protein language modeling.
Design thermostable protein sequences using ProteinMPNN trained on hyperthermophilic organism structures. Generates sequences optimized for improved thermal stability without requiring ligands or additional context.
Design protein sequences for given backbone structures using deep learning. Fast and accurate inverse folding with state-of-the-art sequence recovery (52.4%).
Specialized model for soluble protein sequence design. Trained exclusively on soluble proteins for optimized performance on cytoplasmic and extracellular proteins.
Design antibody heavy- and light-chain CDR sequences from an antibody-antigen complex with the IgDesign inverse-folding model.
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
Inverse folding with ESM-IF1. Design protein sequences for given 3D backbone structures using a geometric deep learning model. Generate multiple sequence variants optimized for your target structure.
ProFam-1 is a protein family language model for family-conditioned sequence generation. Provide a protein family in FASTA, A2M, or A3M format and generate new sequences with model likelihood scores for downstream ranking and screening.
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
All-atom generative diffusion model for protein design with complex constraints. Design binders, enzymes, and symmetric protein assemblies.
Design linear peptide binders for target proteins using a target sequence-conditioned masked language model. PepMLM generates peptide sequences optimized to bind specific protein targets based on ESM-2 protein language modeling.
Configure inputs to begin
Set options on the left, then click “Submit job”.
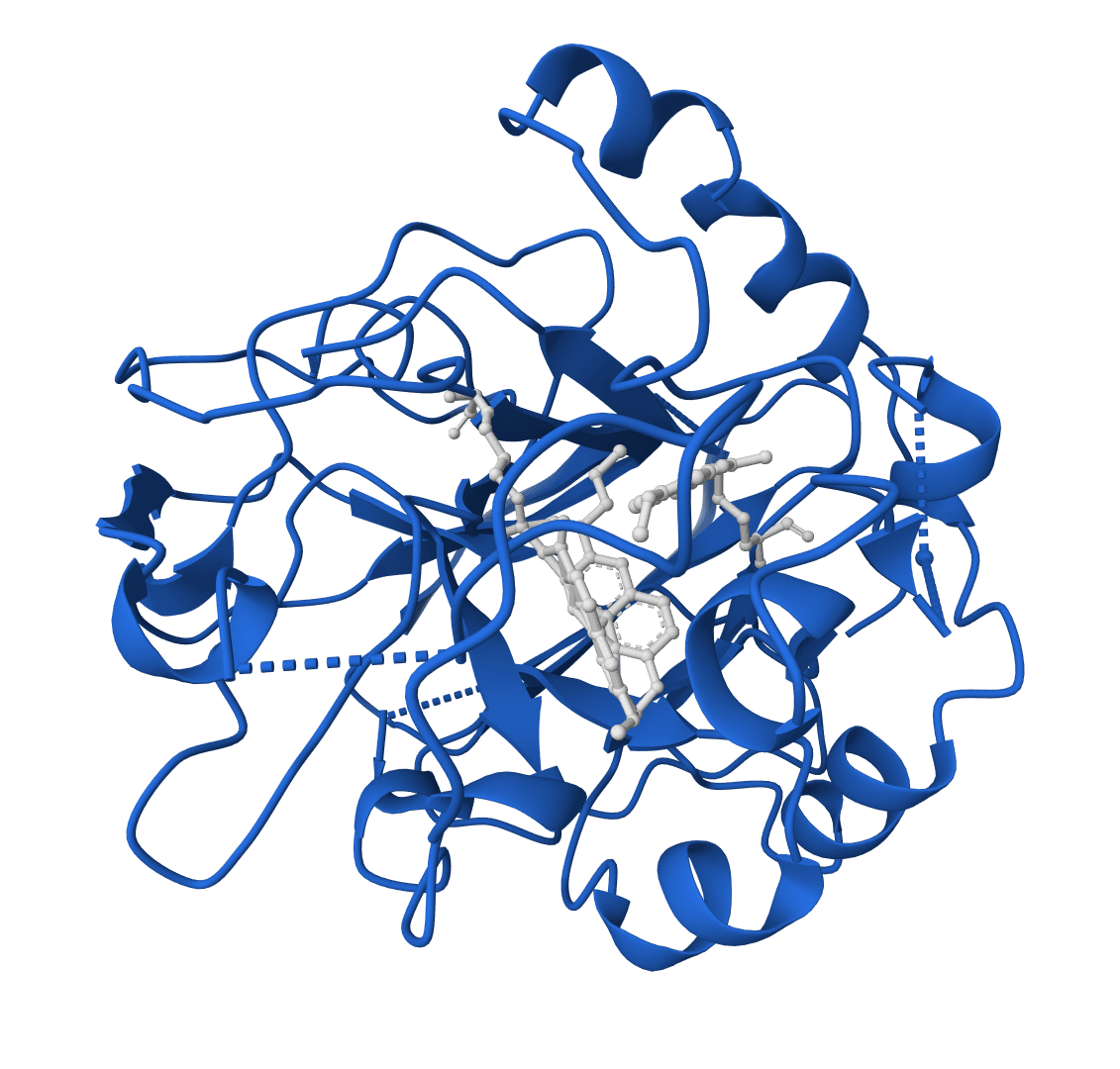
LigandMPNN is an inverse folding model that designs protein sequences while accounting for non-protein atoms—ligands, metals, nucleotides, and cofactors. Standard inverse folding methods like ProteinMPNN only see the protein backbone, which limits their accuracy at binding sites where interactions with small molecules matter most. LigandMPNN solves this by incorporating atomic context from heteroatoms during sequence prediction.
The improvement is substantial. At small molecule binding sites, LigandMPNN achieves 63.3% sequence recovery compared to 50.5% for ProteinMPNN. Metal coordination sites show an even larger gap: 77.5% versus 36.0%. These gains translate to better experimental success rates—over 100 designs have been validated, with some showing 100-fold affinity improvements over conventional methods.
Developed by Dauparas, Lee, and colleagues at the Institute for Protein Design, LigandMPNN was published in Nature Methods (2025).
LigandMPNN extends ProteinMPNN's graph neural network by processing three interconnected graphs instead of one: a protein-only graph (residues as nodes), a ligand-only graph (heteroatoms as nodes), and a protein-ligand graph that captures residue-atom interactions. This architecture allows the model to learn chemical element identities and geometric constraints at binding interfaces.
The model consists of a protein backbone encoder (3 layers), a protein-ligand encoder (2 layers), and an autoregressive sequence decoder. The LigandMPNN source project also includes an optional sidechain-packing model, but ProteinIQ currently returns sequence designs mapped onto the submitted backbone rather than packed full-atom sidechain conformations. Training used 8–16 context atoms per residue and added 0.1 Å Gaussian noise to coordinates, improving generalization to novel structures.
ProteinIQ runs LigandMPNN on managed cloud compute, eliminating the need to install Python dependencies or configure model files locally.
| Input | Description |
|---|---|
Protein | PDB or ENT file (up to 50 MB), or an RCSB PDB ID. Ligands, metals, nucleotides, and cofactors must already be present as HETATM records in this structure. |
LigandMPNN does not accept a separate ligand file in ProteinIQ. If your protein and ligand are stored separately, combine them into one correctly positioned PDB structure before running the design.
| Setting | Description |
|---|---|
Number of sequences | How many sequence variants to generate (1–48, default 1). More sequences give better coverage of sequence space but increase runtime. Use 8–10 for an initial screen or 20–40 for broader exploration. |
Sampling temperature | Positive sampling temperature controlling diversity (default 0.1). Lower values produce conservative designs; values above 1 increasingly flatten the sampling distribution. |
Use atom context | Include ligands, metals, and nucleotides during design (default on). Disabling this makes LigandMPNN behave like ProteinMPNN—only useful as a control. |
Use side chain context | Include fixed residue sidechains as geometric constraints. Enable when catalytic or binding residues are fixed and their geometry should influence neighboring positions. |
Random seed | Integer for reproducible results (0–99999, default 111). Same seed and settings produce identical output. |
| Setting | Description |
|---|---|
Chains to design | Specify which chains to redesign (e.g., A,B); all others stay fixed. Simpler than listing every fixed residue for multi-chain proteins. |
Homo-oligomer | Enable symmetric design for proteins with identical chains. All chains receive the same sequence. |
Fixed positions | Residues to keep unchanged. Format: A15, A1-10, B1-20, or C for an entire chain. Comma-separate multiple entries. |
Redesigned positions | Inverse of fixed positions—specify what to design and fix everything else. Cannot be used simultaneously with Fixed positions. |
Parse chains only | Parse only specified chains from the PDB, ignoring all others. Useful for large assemblies where only a subset of chains is relevant. |
Scoring cutoff (Å) | Distance cutoff for selecting residues scored with ligand context (default 8.0 Å). Decrease to focus design on residues very close to the ligand; increase for broader context. |
Include zero-occupancy atoms | Include atoms with zero occupancy from crystal structures, which indicate partially occupied or disordered positions. Off by default. |
Exclude amino acids | Globally exclude one or more amino acids from all designed positions. Enter one-letter codes without separators (e.g., CW to exclude cysteine and tryptophan). |
Amino acid biases | Adjust sampling frequency per amino acid. Positive values increase likelihood and negative values decrease it. Values near −25 effectively exclude an amino acid; use Exclude amino acids for exact exclusions. |
Each designed sequence includes:
| Column | Description |
|---|---|
Sequence ID | Unique identifier for the design (seq_1, seq_2, …) |
Sequence | Designed amino acid sequence. The native FASTA separates chains with :; the generated-design convenience FASTA uses /. |
Length | Total residue count across all designed chains |
Overall confidence | Model confidence (0–1), computed as the geometric mean probability over redesigned residues. Higher values indicate stronger sequence–structure compatibility. |
Ligand confidence | Confidence specifically at residues within the scoring cutoff distance of ligand atoms. Distinct from overall confidence; provides a focused view of binding site quality. |
Seq recovery | Fraction of redesigned positions matching the input sequence |
Mutation count | Number of positions that differ from the input, using the submitted PDB residue numbers and insertion codes |
Identity % | Percent identity to the input sequence |
Results include the unchanged native LigandMPNN FASTA, a generated-design-only FASTA, a spreadsheet view, native statistics, prediction provenance, and one backbone PDB per generated sequence.
| Feature | ProteinMPNN | LigandMPNN |
|---|---|---|
| Input context | Protein backbone only | Protein + ligands, metals, nucleotides |
| Binding site recovery | 50.5% | 63.3% |
| Metal coordination recovery | 36.0% | 77.5% |
| Nucleotide binding recovery | 35.2% | 50.5% |
| Model size | 1.66M parameters | 2.62M parameters |
| Architecture | Single protein graph | Three-graph (protein, ligand, protein-ligand) |
| Optional source-project sidechain packing | No | Available in the source project; not currently run by ProteinIQ |
| Speed | Faster | ~2× slower |
| Credit cost | 25 | 50 |
Use ProteinMPNN for general protein design where no ligands or cofactors are involved—de novo protein scaffolds, antibody frameworks away from CDRs, or soluble protein cores.
Use LigandMPNN whenever the design involves binding sites: enzyme active sites, cofactor-binding pockets (NAD, FAD, heme), metal coordination spheres, or nucleic acid interfaces. The sequence recovery improvements at these sites translate directly to higher experimental success rates.
Use RFdiffusion 2 before LigandMPNN when the backbone itself needs to be generated rather than only resequenced.