EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
RFdiffusion is a state-of-the-art protein structure generation tool that uses diffusion models to design proteins de novo, create binders, scaffold motifs, and generate symmetric oligomers with atomic precision.
ProGen2 is Salesforce Research's protein language model suite for prompt-based de novo protein sequence generation and bidirectional sequence likelihood scoring.
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
RFdiffusion is a state-of-the-art protein structure generation tool that uses diffusion models to design proteins de novo, create binders, scaffold motifs, and generate symmetric oligomers with atomic precision.
ProGen2 is Salesforce Research's protein language model suite for prompt-based de novo protein sequence generation and bidirectional sequence likelihood scoring.
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
Configure inputs to begin
Set options on the left, then click “Submit job”.
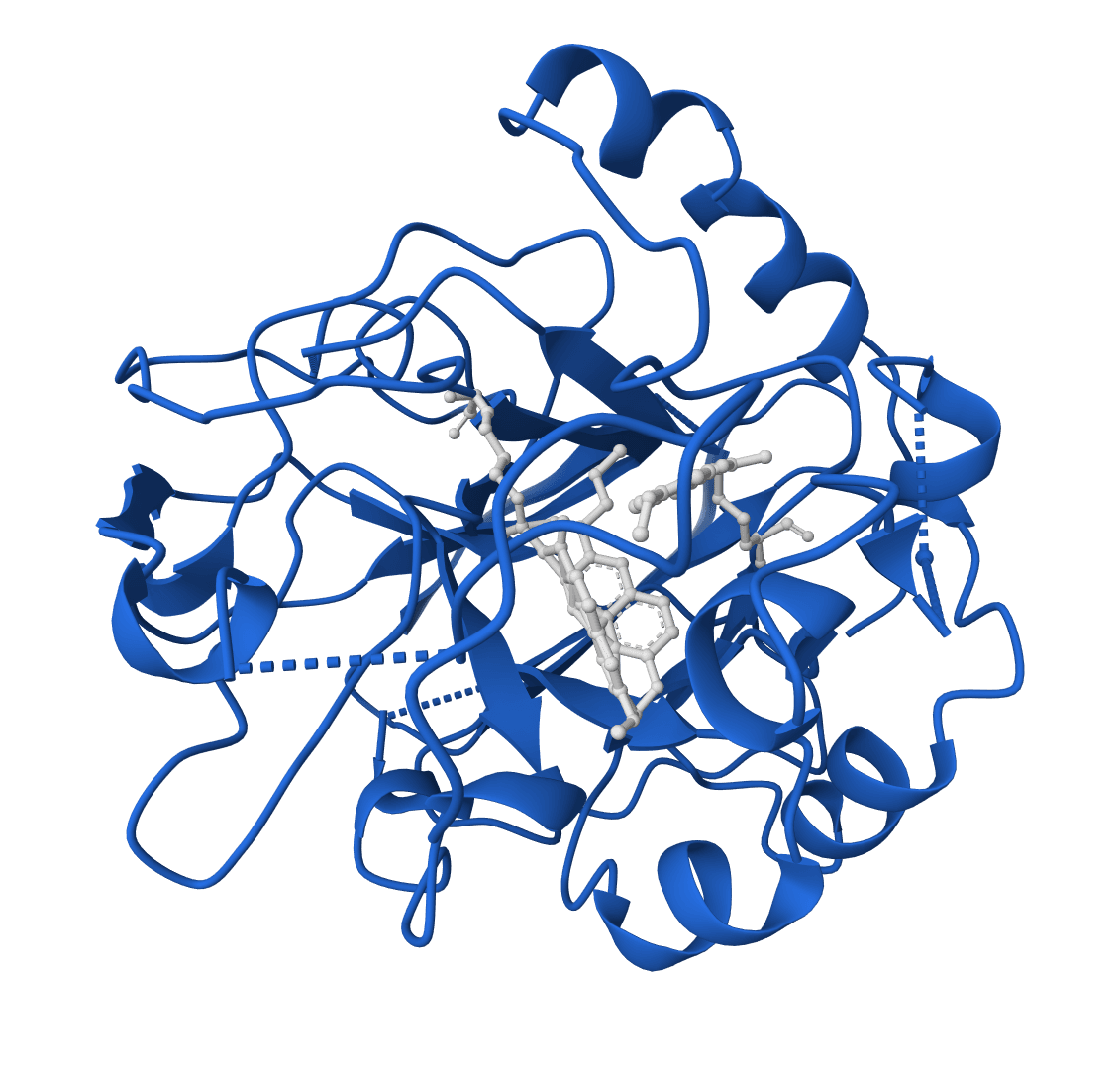
RFdiffusion 2 is a generative deep learning model for enzyme design that scaffolds protein structures around atomic-level descriptions of active sites. Developed by the Baker Lab and the Institute for Protein Design at the University of Washington, it was published in Nature Methods in January 2026. The model builds on RoseTTAFold All-Atom (RFAA) and uses flow matching to generate protein backbones that embed user-specified catalytic geometries, including ligands and metal cofactors, without requiring pre-specified sequence indices or rotamer enumeration.
Where the original RFdiffusion represented active sites at the residue level and required explicit sequence positions for every catalytic residue, RFdiffusion 2 accepts atom-level descriptions of functional groups (known as theozymes) and infers both the residue identity and side-chain conformation during generation. This eliminates the combinatorial explosion of searching over all possible sequence positions and rotamer states, making it practical to scaffold complex multi-residue active sites that were previously intractable.
On a benchmark of 41 diverse enzyme active sites drawn from the Mechanism and Catalytic Site Atlas (M-CSA), RFdiffusion 2 successfully generates scaffolds for all 41 cases, compared to 16 out of 41 for the original RFdiffusion. In experimental validation across five distinct chemical reactions, active enzymes were identified after screening fewer than 96 designs in every case, with the most active zinc metallohydrolase reaching a catalytic efficiency (k_cat/K_M) of 53,000 M^-1 s^-1.
RFdiffusion 2 is designed primarily for de novo enzyme engineering. Typical applications include:
The tool is not intended for general protein binder design or unconditional protein generation. For those tasks, RFdiffusion and RFdiffusion 3 offer broader design modes.
RFdiffusion 2 is built on the RoseTTAFold All-Atom (RFAA) neural network architecture, which can represent residues in two modes simultaneously: as SE(3) backbone frames (N, Ca, C atoms) for standard residues, or as full heavy-atom coordinate sets for "atomized" residues that require side-chain-level precision. During generation, the model processes this mixed-resolution representation and predicts both backbone coordinates and side-chain conformations for atomized positions.
The RFAA base architecture was retrained with creative motif scaffolding tasks. Training used biomolecular structures from the Protein Data Bank, including proteins, protein-small molecule complexes, protein-metal complexes, and covalently modified proteins. Motifs were resampled on each training step to prevent memorization, and the model converged stably over 17 days on 24 NVIDIA A100 GPUs.
Unlike the original RFdiffusion, which used denoising diffusion probabilistic models (DDPM), RFdiffusion 2 employs flow matching. Flow matching interpolates between a noise sample and a training example along a straight path, training the network to predict the original uncorrupted structure from any point along that path. For the SE(3) components of protein frames (rotations and translations), the method uses Riemannian flow matching via the Frame-Flow formulation, removing the approximations for rotational losses that were present in the first RFdiffusion.
Flow matching proved simpler to train: RFdiffusion 2 required no auxiliary losses and no self-conditioning, both of which were necessary for stable training of the original model.
A central innovation in RFdiffusion 2 is the distinction between indexed and unindexed motif residues:
Guidepost mode is the default and recommended setting for most enzyme design tasks. It avoids the combinatorial search over L!/(L-M)! possible assignments of M motif residues to L sequence positions, allowing the model to discover optimal placements naturally during generation.
ORI (origin) tokens provide control over the spatial positioning of the generated scaffold relative to the active site. An ORI pseudo-atom specifies the approximate desired center of mass of the designed protein. During flow matching, a technique called stochastic centering adds a small random global translation so that noised structures encode only an approximate offset between motif and scaffold, allowing the model to refine the positioning across denoising steps.
When the full pipeline is enabled, RFdiffusion 2 feeds generated backbones into LigandMPNN for sequence design. LigandMPNN generates amino acid sequences conditioned on the backbone geometry, catalytic residue side chains, and ligand coordinates. Multiple sequence variants are produced per backbone design to increase the likelihood of finding a foldable, functional sequence. The designed sequences are then evaluated with structure prediction (Chai-1) to verify that they fold to the intended conformation.
ProteinIQ provides GPU-accelerated access to RFdiffusion 2 without local installation, container setup, or command-line configuration.
| Input | Description |
|---|---|
Active site / Motif | PDB file containing the catalytic residues and optionally ligands as HETATM records. Upload a file (.pdb, .cif, .ent) or fetch from RCSB by PDB ID. |
The input structure must contain the active site residues to scaffold. For ligand-aware design, small molecules, metal ions, or cofactors should be included as HETATM records in the PDB file. The model reads both the protein residue coordinates and the ligand atoms to generate scaffolds that accommodate the complete active site geometry.
| Setting | Default | Range | Description |
|---|---|---|---|
Number of samples | 5 | 1-20 | Number of independent scaffold designs to generate. More samples provide structural diversity at the cost of longer computation. |
Run full pipeline | On | On/Off | When on, runs LigandMPNN sequence design after backbone generation. When off, outputs backbone-only structures (faster). |
| Setting | Default | Range | Description |
|---|---|---|---|
N-terminal scaffold | 20 | 0-100 | Number of residues to generate before the motif at the N-terminus. Set to 0 to start directly from the motif. |
C-terminal scaffold | 20 | 0-100 | Number of residues to generate after the motif at the C-terminus. Set to 0 to end at the motif. |
Total scaffold length | 0 (auto) | 0-300 | Target total protein length including motif. When set to a nonzero value, overrides individual N/C-terminal settings. Leave at 0 for automatic sizing. |
| Setting | Default | Range | Description |
|---|---|---|---|
Diffusion steps | 50 | 20-200 | Number of flow matching denoising steps. Higher values may improve structural quality but increase runtime. 50 steps provides a good quality-speed balance. |
Use ORI tokens | Off | On/Off | Enables origin tokens to control the desired center of mass of the scaffold relative to the active site. Useful for controlling where the bulk of the protein sits around the catalytic residues. |
Guidepost mode | On | On/Off | When on, motif positions are treated as flexible guides (unindexed) and the model infers optimal sequence positions. When off, motif positions are fixed at exact sequence indices (indexed). Guidepost mode is recommended for most enzyme design tasks. |
Custom contig | (empty) | Text | Override the auto-generated contig string for advanced control. Format: 20,A1-46,20 where numbers specify scaffold lengths and chain-residue ranges specify preserved motif regions. |
RFdiffusion 2 produces downloadable PDB files for each generated design. When the full pipeline is enabled, output files include LigandMPNN-designed sequences with side chains packed. The 3D viewer displays each design with the scaffolded protein around the preserved active site geometry.
RFdiffusion 2 is not a general replacement for the original RFdiffusion. The two models serve different purposes and have different strengths.
| Feature | RFdiffusion | RFdiffusion 2 |
|---|---|---|
| Primary application | Binder design, motif scaffolding, unconditional generation, symmetric oligomers | Enzyme active site scaffolding |
| Active site representation | Residue-level (backbone frames only) | Atom-level (full side-chain coordinates) |
| Training method | DDPM diffusion with self-conditioning | Flow matching without self-conditioning |
| Architecture base | RoseTTAFold | RoseTTAFold All-Atom (RFAA) |
| Ligand handling | No explicit ligand modeling | Ligands as HETATM with RASA conditioning |
| Sequence position inference | Requires pre-specified indices | Can infer indices via guidepost mode |
| AME benchmark (41 active sites) | 16/41 successful | 41/41 successful |
| Design modes | 5 modes (binder, scaffolding, partial diffusion, unconditional, custom) | Enzyme active site scaffolding with optional ligand conditioning |
| Sequence design | ProteinMPNN + AlphaFold2 validation | LigandMPNN + Chai-1 validation |
For general protein design tasks such as binder design, partial diffusion, unconditional generation, or symmetric oligomer assembly, the original RFdiffusion remains the appropriate tool. For all-atom biomolecular interaction design across proteins, DNA, and small molecules, RFdiffusion 3 extends the approach further.
Each output PDB file represents an independent scaffold design that embeds the input active site geometry. Key aspects to evaluate:
The most predictive metric for experimental success is agreement between the designed structure and the independently predicted structure from the designed sequence. Designs where the predicted structure closely recapitulates the catalytic geometry have the highest probability of exhibiting enzymatic activity.
RFdiffusion 2 is specialized for enzyme active site scaffolding. It does not support binder design, unconditional protein generation, or symmetric oligomer assembly. For those tasks, use RFdiffusion or RFdiffusion 3.
Designed enzymes generally exhibit lower catalytic activity than natural enzymes. Theozyme descriptions may not capture all the interactions required for high activity, such as remote residues that contribute to transition-state stabilization through electrostatic preorganization or dynamic effects. The best experimentally validated designs reach k_cat/K_M values in the tens of thousands (M^-1 s^-1), which is orders of magnitude below typical natural enzymes (10^6 to 10^8 M^-1 s^-1).
Sequence design for regions outside the active site is performed independently by LigandMPNN rather than co-designed with the backbone. Simultaneous backbone and sequence optimization could improve results but is not yet implemented.
The model was trained on structures from the Protein Data Bank, which introduces biases toward well-characterized folds and catalytic mechanisms. Novel chemistries or unusual coordination geometries that are underrepresented in the training data may produce lower-quality scaffolds.