BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
Design protein binders against a target structure with NVIDIA BioNeMo's Proteina-Complexa generative pipeline.
ProFam-1 is a protein family language model for family-conditioned sequence generation. Provide a protein family in FASTA, A2M, or A3M format and generate new sequences with model likelihood scores for downstream ranking and screening.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
Structure-based de novo antibody and nanobody design pipeline combining antibody-tuned RFdiffusion, ProteinMPNN sequence design, and antibody-tuned RoseTTAFold2 filtering.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
Design protein binders against a target structure with NVIDIA BioNeMo's Proteina-Complexa generative pipeline.
ProFam-1 is a protein family language model for family-conditioned sequence generation. Provide a protein family in FASTA, A2M, or A3M format and generate new sequences with model likelihood scores for downstream ranking and screening.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
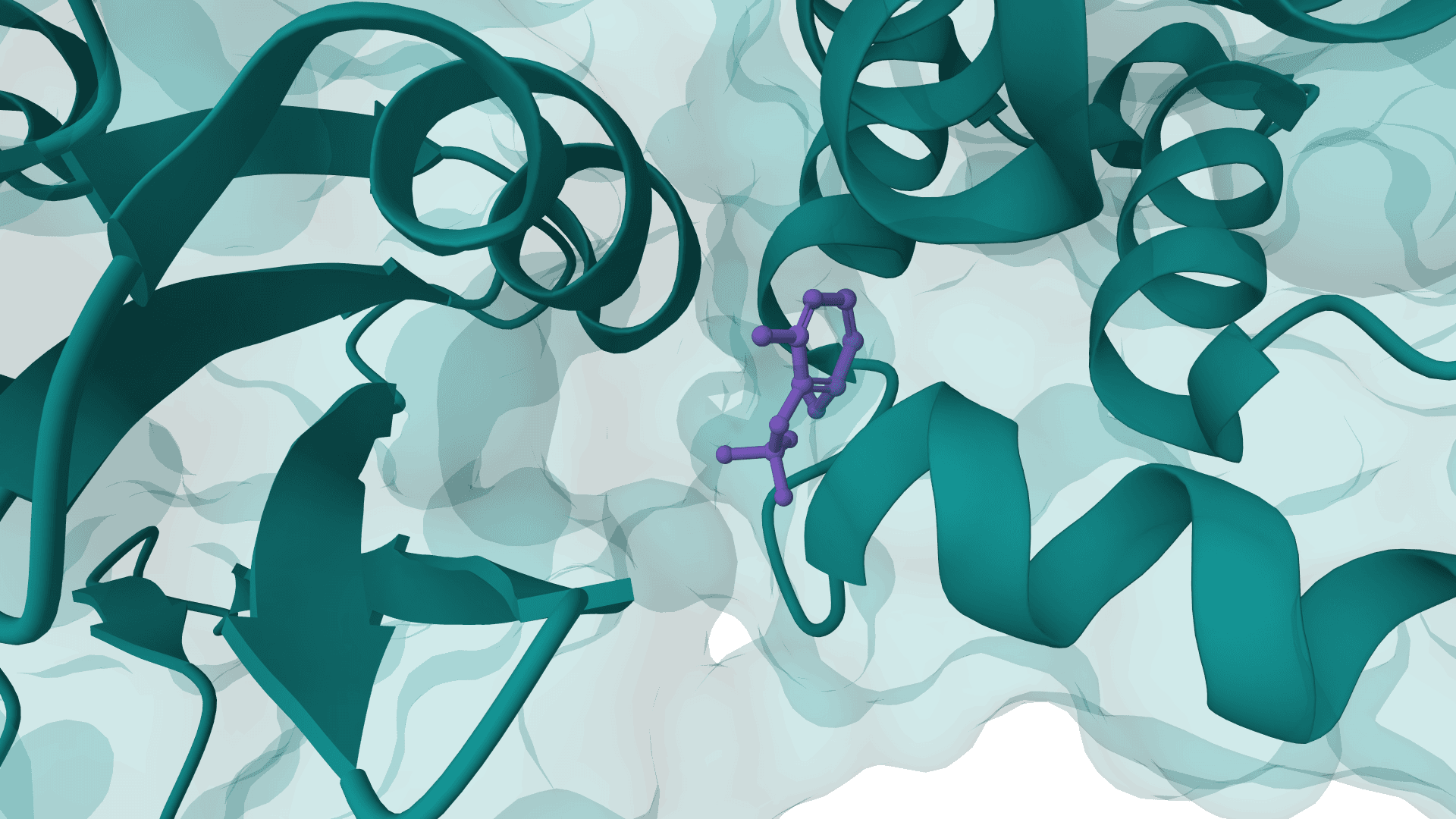
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
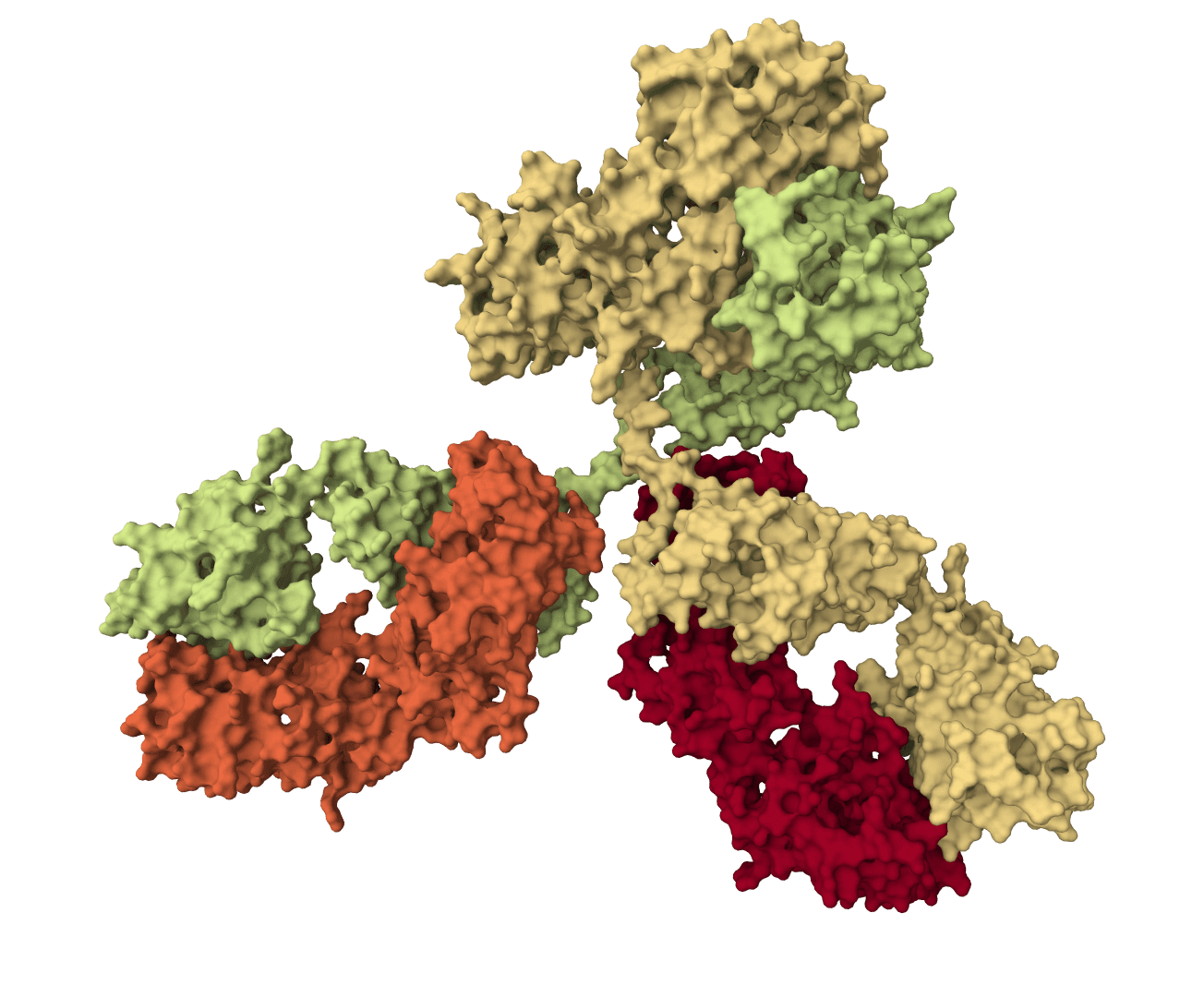
Structure-based de novo antibody and nanobody design pipeline combining antibody-tuned RFdiffusion, ProteinMPNN sequence design, and antibody-tuned RoseTTAFold2 filtering.
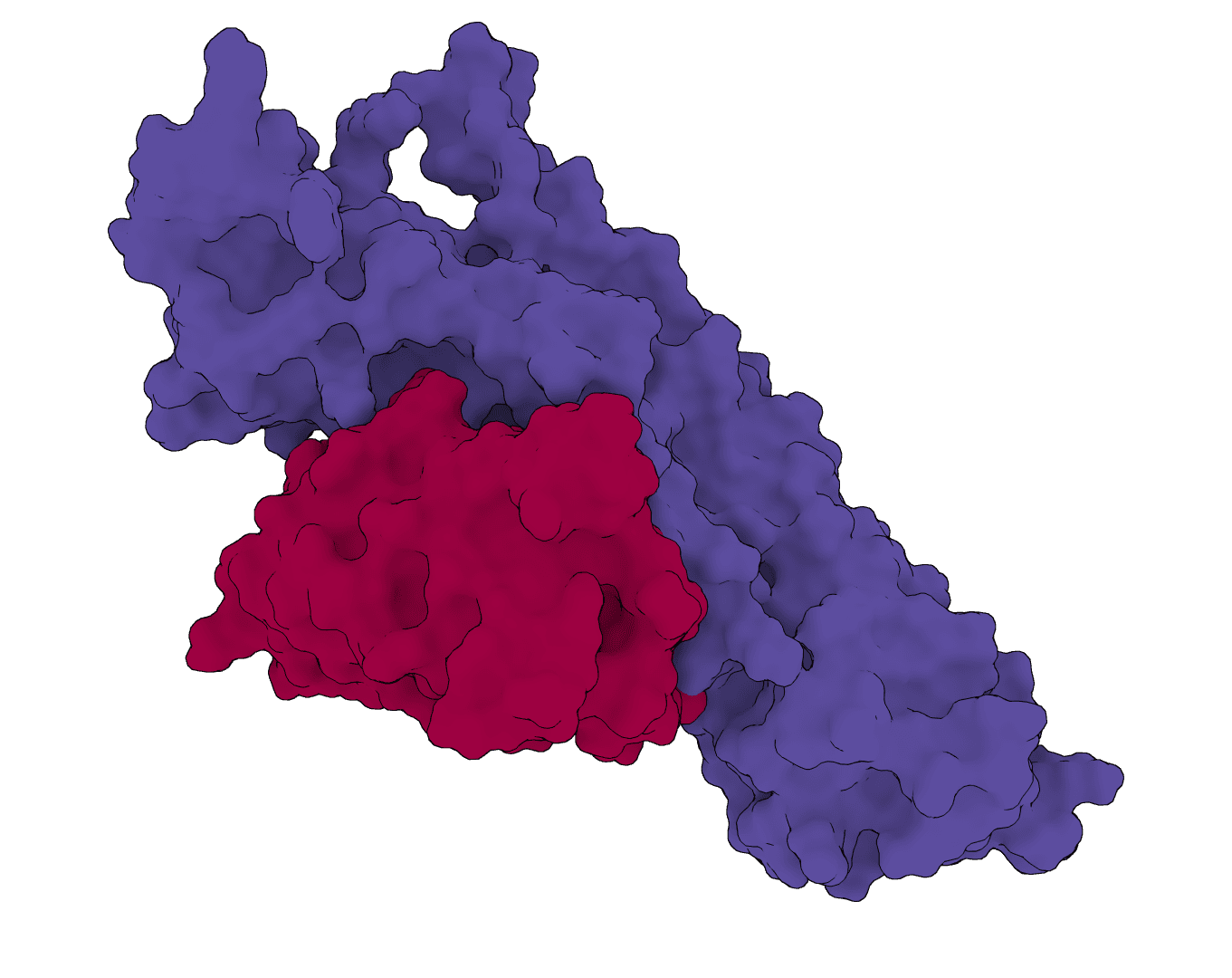
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Configure inputs to begin
Set options on the left, then click “Submit job”.
PepMLM is a de novo peptide binder design tool that generates linear peptide sequences predicted to bind specific target proteins. Developed by the Chatterjee Lab and published in Nature Biotechnology in 2025, PepMLM employs a target sequence-conditioned masked language modeling approach built on ESM-2, a state-of-the-art protein language model.
Unlike structure-based design methods that require 3D coordinates of the target protein, PepMLM operates exclusively from amino acid sequences. The model positions peptide binders at the C-terminus of target proteins and reconstructs the binder region through span masking, enabling generative design of candidate binders to any target protein without structural information.
PepMLM was experimentally validated through binding assays and targeted protein degradation studies, demonstrating efficacy against cancer biomarkers, viral phosphoproteins, and Huntington's disease-related proteins.
ProteinIQ provides a web-based interface for running PepMLM without command-line installation or GPU configuration. Paste or upload target protein sequences, adjust generation parameters, and receive peptide binder candidates with pseudo-perplexity scores.
| Input | Description |
|---|---|
Target Protein Sequence(s) | Amino acid sequence of the target protein. Accepts FASTA format, raw text sequences, or UniProt IDs. Multiple targets can be processed in a single job. Maximum 50 sequences per job and 500 residues per target. Large batches may require fewer binders or a shorter peptide length to stay within one GPU job. |
| Setting | Description |
|---|---|
Peptide length | Length of generated binders in amino acids (3–50, default 15). |
Binders per target | Number of candidate peptides to generate for each target sequence (1–32, default 4). Higher values provide greater sequence diversity but increase computation time. |
Top-k sampling | Restricts sampling to the top most probable amino acids at each position (1–10, default 3). Lower values (1–2) produce conservative, high-confidence designs. Higher values (5–10) increase sequence diversity by exploring more of the probability distribution. |
The output presents peptide binder candidates in generation order and includes the source-compatible output.csv file.
| Column | Description |
|---|---|
Binder | Designed peptide sequence in single-letter amino acid code. Sequences may contain X (any amino acid) at positions where the model assigns similar probabilities to multiple residues. |
Pseudo Perplexity | Model-consistency score for the generated sequence conditioned on the target. Lower values indicate greater consistency with the model, but do not directly predict binding affinity. |
Input Sequence | Full target protein sequence. Only returned when processing multiple targets in one job. |
Pseudo-perplexity quantifies how "surprised" the model is by the generated peptide sequence conditioned on the target protein. The metric is calculated on the binder region by masking one binder token at a time and measuring reconstruction loss.
Use lower values to compare candidates generated for the same target with the same settings. PepMLM does not define universal validated pseudo-perplexity thresholds, and the score is not a direct measurement of binding affinity or experimental success.
PepMLM adapts the ESM-2 protein language model for conditional peptide generation through a novel masking strategy. The approach treats binder design as a sequence reconstruction problem where peptides are predicted given their target protein context.
The core innovation positions peptide sequences at the C-terminus of target proteins during training. The entire binder region is masked, forcing the model to reconstruct it based solely on the target sequence context. This contrasts with traditional masked language models that mask random tokens throughout the sequence.
During inference, the model receives a target protein sequence concatenated with a masked peptide template of length . Top-k sampling with categorical probability distributions generates amino acids position-by-position, producing diverse binder candidates rather than a single deterministic prediction.
PepMLM fine-tunes ESM-2 (650M parameters) on merged datasets from PepNN and Propedia, comprising 10,000 training samples and 203 test sequences. The model constrains binders to maximum 50 amino acids and target proteins to 500 residues.
The training objective minimizes masked language modeling loss exclusively on binder regions:
where represents amino acids in the masked binder region, is the target protein sequence, and includes already-generated positions.
In comparative studies, PepMLM achieved superior performance against RFdiffusion on structured targets:
X (any amino acid), requiring manual substitution or experimental screening