RFdiffusion is a generative AI model for designing novel protein structures with atomic precision. Developed by the Baker Lab at the University of Washington and published in Nature (2023), it uses diffusion models—the same technology behind DALL-E and Stable Diffusion—adapted for protein structure generation. With over 1,000 citations since publication, the method achieves approximately two orders of magnitude improvement over traditional computational protein design approaches.
Independent experimental validation demonstrates 84% confirmation rates for designed binders, with affinities ranging from tens of micromolar to tens of nanomolar. Applications include therapeutic binder design, enzyme active site scaffolding, symmetric oligomer creation, and de novo protein generation.
RFdiffusion works by iteratively denoising random 3D coordinates into structured protein backbones through learned reverse diffusion steps. Unlike traditional methods that optimize physics-based energy functions, it learns directly from the statistical distribution of natural protein structures in the Protein Data Bank.
RFdiffusion builds on the RoseTTAFold (RF) structure prediction network architecture, similar to AlphaFold2. The model was initialized with RoseTTAFold's pretrained weights and fine-tuned specifically for structure denoising tasks. It uses SE(3)-equivariant graph neural networks that respect rotational and translational symmetries, improving generalization and data efficiency.
The architecture processes protein structures as rigid-frame representations—coordinates and orientations of backbone atoms (N, Cα, C, O, and virtual Cβ). This representation enables precise control at the residue level while maintaining physical realism.
The model operates through reverse diffusion on protein backbone structures. Starting from random noise (random 3D coordinates), it iteratively denoises through learned steps toward a structured protein. At each timestep, the model predicts the final structure from the current noised structure, then interpolates from the current coordinates toward the predicted structure.
Training used 200 discrete diffusion timesteps, though the default deployment uses 50 steps for optimal quality-speed balance. Research shows that as few as 20 timesteps achieve equivalent quality with 10x speedup. Higher timestep counts improve quality with diminishing returns beyond 50.
A critical innovation is self-conditioning: the model receives its previous prediction as template input, similar to AlphaFold2's "recycling" mechanism. This architectural choice improves prediction quality by allowing the model to iteratively refine its understanding of the emerging structure.
Unlike traditional protein design methods requiring manual parameter tuning and physics-based optimization, RFdiffusion treats design as generative modeling over protein structure space. It requires no evolutionary information or multiple sequence alignments (MSAs). The approach enables diverse design tasks through conditional generation—specifying constraints like binding sites, motifs, or symmetry while allowing the model to generate compatible structures.
RFdiffusion supports five distinct design modes for different applications:
Creates proteins that bind to a specified target protein. You provide the target structure and optionally specify binding pocket residues (hotspots). The model generates novel binder proteins with specified length ranges that form interfaces with the target. This mode has been experimentally validated with picomolar-affinity binders to therapeutic targets including MDM2, PD-L1, and IL-7Rα.
Builds a protein scaffold around a functional motif of interest. You specify which residues to preserve and how much structure to add at the N- and C-termini. Applications include scaffolding viral epitopes, receptor binding sites, enzyme active sites, and metal-binding motifs. This enables transplanting functional elements into new structural contexts.
Partially redesigns an existing protein structure to create variants. By controlling the diffusion timesteps (partial_T), you can tune the diversity—low values create subtle variations, high values generate more radical changes. Useful for exploring structural space around a known fold while maintaining key geometric features.
Generates novel protein structures from scratch without any template. You specify the desired length and optional symmetry. The model samples entirely new protein folds from learned structural distributions. This mode enables exploring uncharted regions of protein structure space.
Advanced mode for users who want precise control using contig syntax. Contigs define exactly which regions to keep from input structures and where to generate new structure. This enables complex design tasks like inserting domains, creating fusion proteins, or specifying exact topological arrangements.
RFdiffusion accepts PDB format structures (.pdb, .ent files) up to 50 MB. You can upload local files or provide RCSB PDB IDs to fetch structures directly. Most modes require an input structure, except unconditional generation which starts from scratch.
A10-100/0 50-150 keeps A10-100, breaks chain, generates 50-150 residue chain. Advanced users requiring exact topological specifications. See RFdiffusion GitHub for full syntax.RFdiffusion returns its native backbone design files:
Xt-1 and pX0 multi-step trajectory files when trajectory output is enabled by RFdiffusion. These show the denoising path and can be inspected in molecular viewers that support multi-model PDBs.Run ProteinMPNN, LigandMPNN, AlphaFold, or a related validation workflow after RFdiffusion when you need sequence design, side-chain packing, or fold-confidence metrics.
High-affinity binders to therapeutic targets represent the most experimentally validated application. Published examples include nanomolar binders to MDM2 (0.5-0.7 nM vs 600 nM native), influenza hemagglutinin, IL-7Rα, PD-L1, and TrkA. Independent validation by Adaptyv Bio on IL-7Rα demonstrated 84% confirmation rate with 23/27 binders showing measurable affinity (strongest: 40 nM).
Cryo-EM structures of designed binders show near-perfect agreement with computational models. Some designed interfaces achieve picomolar affinity through pure computation without experimental optimization.
RFdiffusion can scaffold catalytic residues into novel protein folds with specified symmetry. The method enables de novo enzyme design by building NTF2-like folds around functional sites. Applications include designing protein-metal assemblies around coordination sites and transferring catalytic motifs into alternative structural contexts.
Generates novel symmetric assemblies validated by electron microscopy across all symmetry types—cyclic (Cn), dihedral (Dn), tetrahedral (T), octahedral (O), and icosahedral (I). Examples include C3 symmetric trimers targeting SARS-CoV-2 spike protein. Hundreds of designed metal-binding symmetric proteins have been experimentally characterized.
Unconditional mode creates entirely novel protein folds not seen in nature. No template or evolutionary information required. Topology-constrained monomer designs demonstrate the model's ability to explore uncharted regions of protein structure space.
Original Nature paper: 55/96 designs (57%) showed detectable binding at 10 μM, representing ~2 orders of magnitude improvement over previous methods. However, recent critical evaluations show variable success for challenging eukaryotic targets, highlighting the need for generating hundreds to thousands of designs for difficult cases.
Newer methods like BindCraft and EvoPro achieve ~50% success rates (>10x improvement over earlier approaches), while specialized tools show varying performance: RFpeptides (macrocycles) 1.72%, Latent-X 8.26%.
Start with unconditional mode (100-150 residues) to understand outputs and quality metrics. Use default parameters initially: 10 designs, 50 timesteps, standard temperature settings. Begin with simple design tasks before adding guiding potentials or complex contigs.
Define motif precisely with exact residue ranges. Allow sufficient N/C-terminal extensions (minimum 5-15 residues) for structural stability. Use substrate_contacts guiding potential if ligand is present. Validate that motif geometry is preserved in outputs by measuring RMSD.
Start with partial_T=25-30 for moderate diversity around existing fold. Increase to 40-50 only when more radical structural changes are needed. Use provide_seq parameter to retain sequences of critical functional residues. Check RMSD to input structure to ensure appropriate diversity level.
Increase timesteps when facing complex topology requirements, poor initial results at 50 steps, or preparing final production runs. Enable beta model when outputs show excessive helices or need better secondary structure balance. Add guiding potentials only after trying without them first—use for specific biophysical requirements informed by experimental feedback.
Skipping downstream fold-confidence checks can hide weak designs. Testing only the rank 1 design misses diversity—examine top 3-5. Insufficient sampling (too few designs) reduces statistical power for finding successful candidates. Skipping proper protein preparation leads to artifacts. Over-reliance on any single metric misses important failure modes captured by complementary scores.
Standard RFdiffusion performs backbone-only design without explicit side chain or ligand modeling (use LigandMPNN for post-processing). The target protein is treated as rigid during design. Water molecules and explicit solvation are not modeled. Computational cost requires a 20-minute inference timeout and 250 credits per job on ProteinIQ.
Success rates vary from 1-50% depending on target difficulty. Designed sequences may show low recombinant expression in standard systems. Some designs exhibit non-specific binding or promiscuity. Affinity ranges cluster in hundreds of nanomolar, often requiring optimization for therapeutic applications.
Primarily designs with canonical amino acids. Practical size constraints: 10-500 residues. Novel fold generation remains challenging despite improvements over alternatives. Metalloproteins require special consideration. Covalent modifications are not directly supported.
Confidence scores are predictions, not guarantees of experimental success. Training on x-ray crystal structures may reduce performance on computational models. Domain generalization struggles with highly novel protein families. Limited training data for macrocycles and non-standard protein architectures.
ProteinIQ pricing: 250 credits base cost per job. 20-minute inference timeout limit. Moderate-to-high cost tool reflecting complex AI model and GPU-intensive computation.
Optimization: Start with 10 designs (not 50), use 50 timesteps (not 200), test small-scale before large campaigns, batch related designs together.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
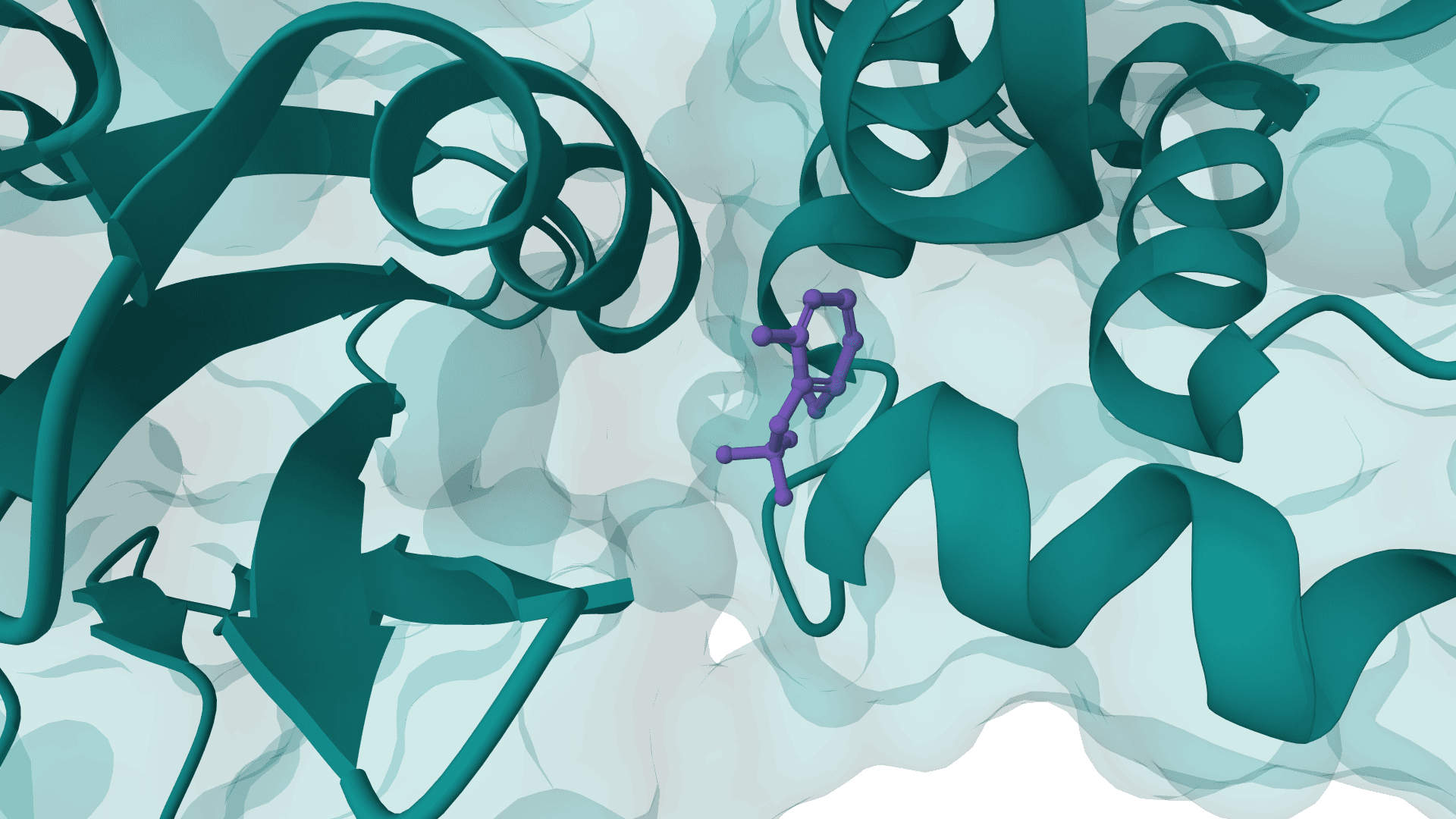
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
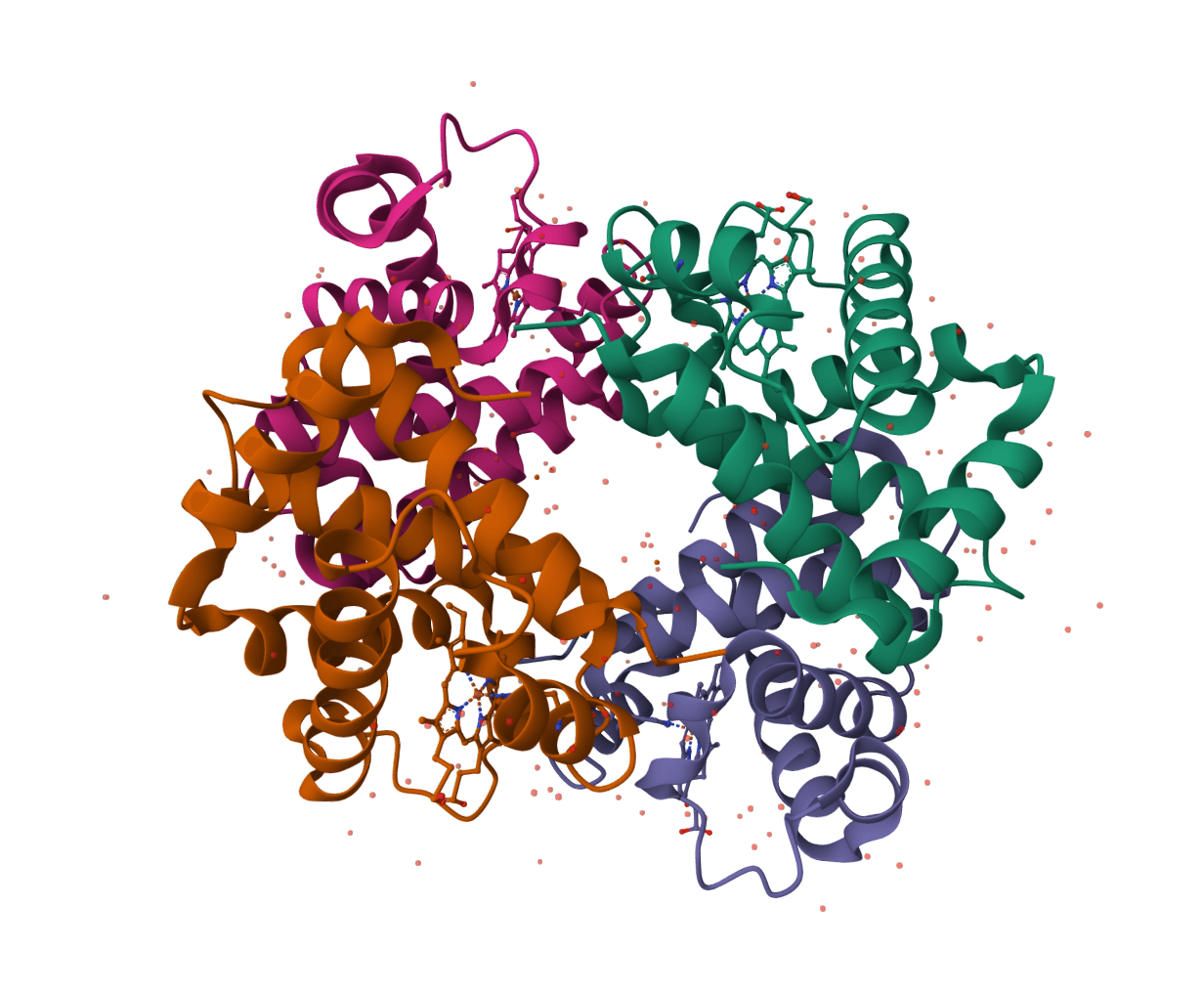
PocketXMol is a pocket-interacting generative foundation model for docking, small-molecule design, and peptide design in protein binding pockets.
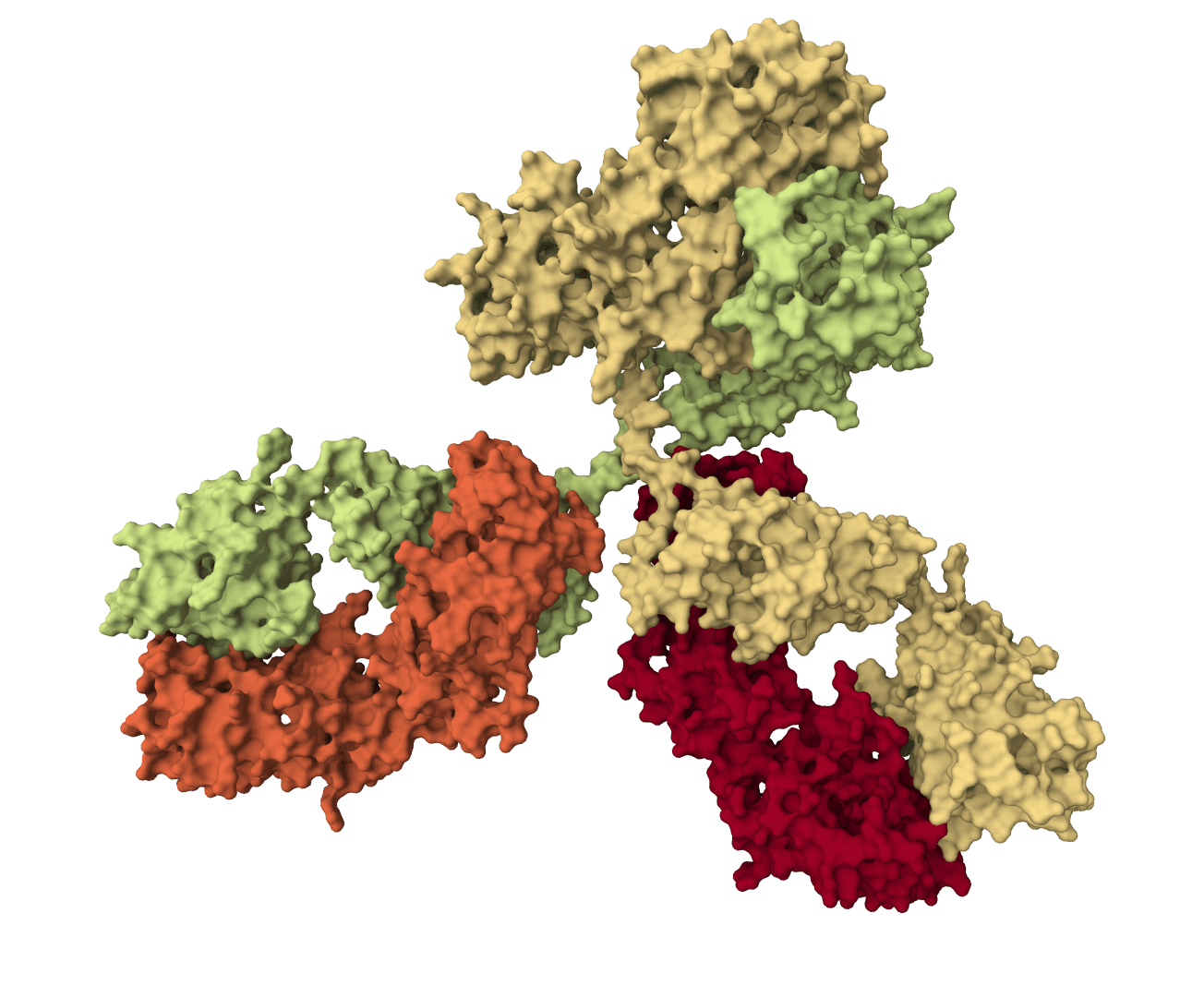
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
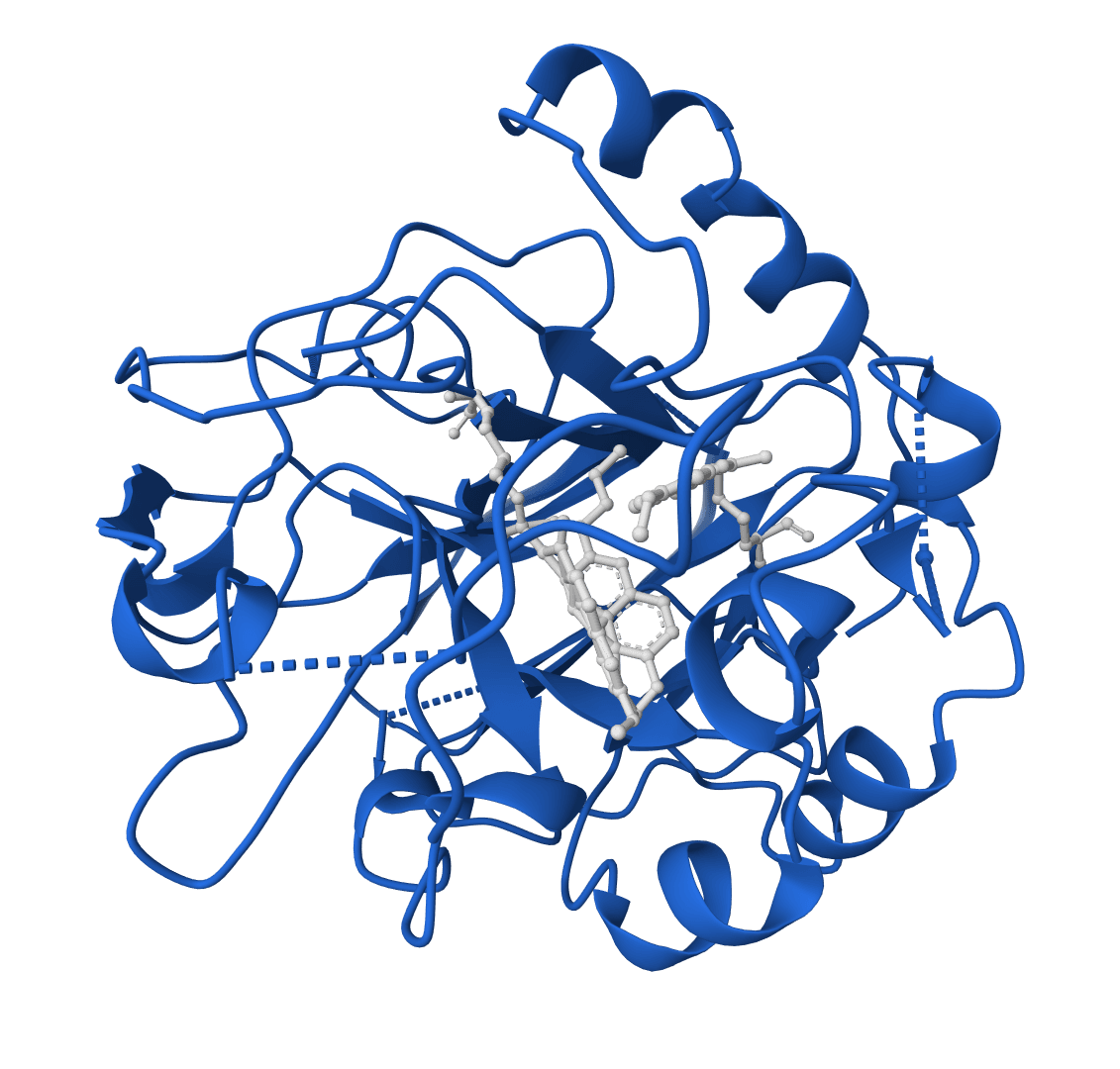
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
ProGen2 is Salesforce Research's protein language model suite for prompt-based de novo protein sequence generation and bidirectional sequence likelihood scoring.
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
RFdiffusion is a generative AI model for designing novel protein structures with atomic precision. Developed by the Baker Lab at the University of Washington and published in Nature (2023), it uses diffusion models—the same technology behind DALL-E and Stable Diffusion—adapted for protein structure generation. With over 1,000 citations since publication, the method achieves approximately two orders of magnitude improvement over traditional computational protein design approaches.
Independent experimental validation demonstrates 84% confirmation rates for designed binders, with affinities ranging from tens of micromolar to tens of nanomolar. Applications include therapeutic binder design, enzyme active site scaffolding, symmetric oligomer creation, and de novo protein generation.
RFdiffusion works by iteratively denoising random 3D coordinates into structured protein backbones through learned reverse diffusion steps. Unlike traditional methods that optimize physics-based energy functions, it learns directly from the statistical distribution of natural protein structures in the Protein Data Bank.
RFdiffusion builds on the RoseTTAFold (RF) structure prediction network architecture, similar to AlphaFold2. The model was initialized with RoseTTAFold's pretrained weights and fine-tuned specifically for structure denoising tasks. It uses SE(3)-equivariant graph neural networks that respect rotational and translational symmetries, improving generalization and data efficiency.
The architecture processes protein structures as rigid-frame representations—coordinates and orientations of backbone atoms (N, Cα, C, O, and virtual Cβ). This representation enables precise control at the residue level while maintaining physical realism.
The model operates through reverse diffusion on protein backbone structures. Starting from random noise (random 3D coordinates), it iteratively denoises through learned steps toward a structured protein. At each timestep, the model predicts the final structure from the current noised structure, then interpolates from the current coordinates toward the predicted structure.
Training used 200 discrete diffusion timesteps, though the default deployment uses 50 steps for optimal quality-speed balance. Research shows that as few as 20 timesteps achieve equivalent quality with 10x speedup. Higher timestep counts improve quality with diminishing returns beyond 50.
A critical innovation is self-conditioning: the model receives its previous prediction as template input, similar to AlphaFold2's "recycling" mechanism. This architectural choice improves prediction quality by allowing the model to iteratively refine its understanding of the emerging structure.
Unlike traditional protein design methods requiring manual parameter tuning and physics-based optimization, RFdiffusion treats design as generative modeling over protein structure space. It requires no evolutionary information or multiple sequence alignments (MSAs). The approach enables diverse design tasks through conditional generation—specifying constraints like binding sites, motifs, or symmetry while allowing the model to generate compatible structures.
RFdiffusion supports five distinct design modes for different applications:
Creates proteins that bind to a specified target protein. You provide the target structure and optionally specify binding pocket residues (hotspots). The model generates novel binder proteins with specified length ranges that form interfaces with the target. This mode has been experimentally validated with picomolar-affinity binders to therapeutic targets including MDM2, PD-L1, and IL-7Rα.
Builds a protein scaffold around a functional motif of interest. You specify which residues to preserve and how much structure to add at the N- and C-termini. Applications include scaffolding viral epitopes, receptor binding sites, enzyme active sites, and metal-binding motifs. This enables transplanting functional elements into new structural contexts.
Partially redesigns an existing protein structure to create variants. By controlling the diffusion timesteps (partial_T), you can tune the diversity—low values create subtle variations, high values generate more radical changes. Useful for exploring structural space around a known fold while maintaining key geometric features.
Generates novel protein structures from scratch without any template. You specify the desired length and optional symmetry. The model samples entirely new protein folds from learned structural distributions. This mode enables exploring uncharted regions of protein structure space.
Advanced mode for users who want precise control using contig syntax. Contigs define exactly which regions to keep from input structures and where to generate new structure. This enables complex design tasks like inserting domains, creating fusion proteins, or specifying exact topological arrangements.
RFdiffusion accepts PDB format structures (.pdb, .ent files) up to 50 MB. You can upload local files or provide RCSB PDB IDs to fetch structures directly. Most modes require an input structure, except unconditional generation which starts from scratch.
A10-100/0 50-150 keeps A10-100, breaks chain, generates 50-150 residue chain. Advanced users requiring exact topological specifications. See RFdiffusion GitHub for full syntax.RFdiffusion returns its native backbone design files:
Xt-1 and pX0 multi-step trajectory files when trajectory output is enabled by RFdiffusion. These show the denoising path and can be inspected in molecular viewers that support multi-model PDBs.Run ProteinMPNN, LigandMPNN, AlphaFold, or a related validation workflow after RFdiffusion when you need sequence design, side-chain packing, or fold-confidence metrics.
High-affinity binders to therapeutic targets represent the most experimentally validated application. Published examples include nanomolar binders to MDM2 (0.5-0.7 nM vs 600 nM native), influenza hemagglutinin, IL-7Rα, PD-L1, and TrkA. Independent validation by Adaptyv Bio on IL-7Rα demonstrated 84% confirmation rate with 23/27 binders showing measurable affinity (strongest: 40 nM).
Cryo-EM structures of designed binders show near-perfect agreement with computational models. Some designed interfaces achieve picomolar affinity through pure computation without experimental optimization.
RFdiffusion can scaffold catalytic residues into novel protein folds with specified symmetry. The method enables de novo enzyme design by building NTF2-like folds around functional sites. Applications include designing protein-metal assemblies around coordination sites and transferring catalytic motifs into alternative structural contexts.
Generates novel symmetric assemblies validated by electron microscopy across all symmetry types—cyclic (Cn), dihedral (Dn), tetrahedral (T), octahedral (O), and icosahedral (I). Examples include C3 symmetric trimers targeting SARS-CoV-2 spike protein. Hundreds of designed metal-binding symmetric proteins have been experimentally characterized.
Unconditional mode creates entirely novel protein folds not seen in nature. No template or evolutionary information required. Topology-constrained monomer designs demonstrate the model's ability to explore uncharted regions of protein structure space.
Original Nature paper: 55/96 designs (57%) showed detectable binding at 10 μM, representing ~2 orders of magnitude improvement over previous methods. However, recent critical evaluations show variable success for challenging eukaryotic targets, highlighting the need for generating hundreds to thousands of designs for difficult cases.
Newer methods like BindCraft and EvoPro achieve ~50% success rates (>10x improvement over earlier approaches), while specialized tools show varying performance: RFpeptides (macrocycles) 1.72%, Latent-X 8.26%.
Start with unconditional mode (100-150 residues) to understand outputs and quality metrics. Use default parameters initially: 10 designs, 50 timesteps, standard temperature settings. Begin with simple design tasks before adding guiding potentials or complex contigs.
Define motif precisely with exact residue ranges. Allow sufficient N/C-terminal extensions (minimum 5-15 residues) for structural stability. Use substrate_contacts guiding potential if ligand is present. Validate that motif geometry is preserved in outputs by measuring RMSD.
Start with partial_T=25-30 for moderate diversity around existing fold. Increase to 40-50 only when more radical structural changes are needed. Use provide_seq parameter to retain sequences of critical functional residues. Check RMSD to input structure to ensure appropriate diversity level.
Increase timesteps when facing complex topology requirements, poor initial results at 50 steps, or preparing final production runs. Enable beta model when outputs show excessive helices or need better secondary structure balance. Add guiding potentials only after trying without them first—use for specific biophysical requirements informed by experimental feedback.
Skipping downstream fold-confidence checks can hide weak designs. Testing only the rank 1 design misses diversity—examine top 3-5. Insufficient sampling (too few designs) reduces statistical power for finding successful candidates. Skipping proper protein preparation leads to artifacts. Over-reliance on any single metric misses important failure modes captured by complementary scores.
Standard RFdiffusion performs backbone-only design without explicit side chain or ligand modeling (use LigandMPNN for post-processing). The target protein is treated as rigid during design. Water molecules and explicit solvation are not modeled. Computational cost requires a 20-minute inference timeout and 250 credits per job on ProteinIQ.
Success rates vary from 1-50% depending on target difficulty. Designed sequences may show low recombinant expression in standard systems. Some designs exhibit non-specific binding or promiscuity. Affinity ranges cluster in hundreds of nanomolar, often requiring optimization for therapeutic applications.
Primarily designs with canonical amino acids. Practical size constraints: 10-500 residues. Novel fold generation remains challenging despite improvements over alternatives. Metalloproteins require special consideration. Covalent modifications are not directly supported.
Confidence scores are predictions, not guarantees of experimental success. Training on x-ray crystal structures may reduce performance on computational models. Domain generalization struggles with highly novel protein families. Limited training data for macrocycles and non-standard protein architectures.
ProteinIQ pricing: 250 credits base cost per job. 20-minute inference timeout limit. Moderate-to-high cost tool reflecting complex AI model and GPU-intensive computation.
Optimization: Start with 10 designs (not 50), use 50 timesteps (not 200), test small-scale before large campaigns, batch related designs together.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
PocketXMol is a pocket-interacting generative foundation model for docking, small-molecule design, and peptide design in protein binding pockets.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
ProGen2 is Salesforce Research's protein language model suite for prompt-based de novo protein sequence generation and bidirectional sequence likelihood scoring.
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.