Design all-atom protein binders with controlled target sites, lengths, and interaction parameters. Learn more
Design all-atom protein binders with controlled target sites, lengths, and interaction parameters. Learn more
Design all-atom protein binders with controlled target sites, lengths, and interaction parameters. Learn more
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
ProGen2 is Salesforce Research's protein language model suite for prompt-based de novo protein sequence generation and bidirectional sequence likelihood scoring.
ProFam-1 is a protein family language model for family-conditioned sequence generation. Provide a protein family in FASTA, A2M, or A3M format and generate new sequences with model likelihood scores for downstream ranking and screening.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
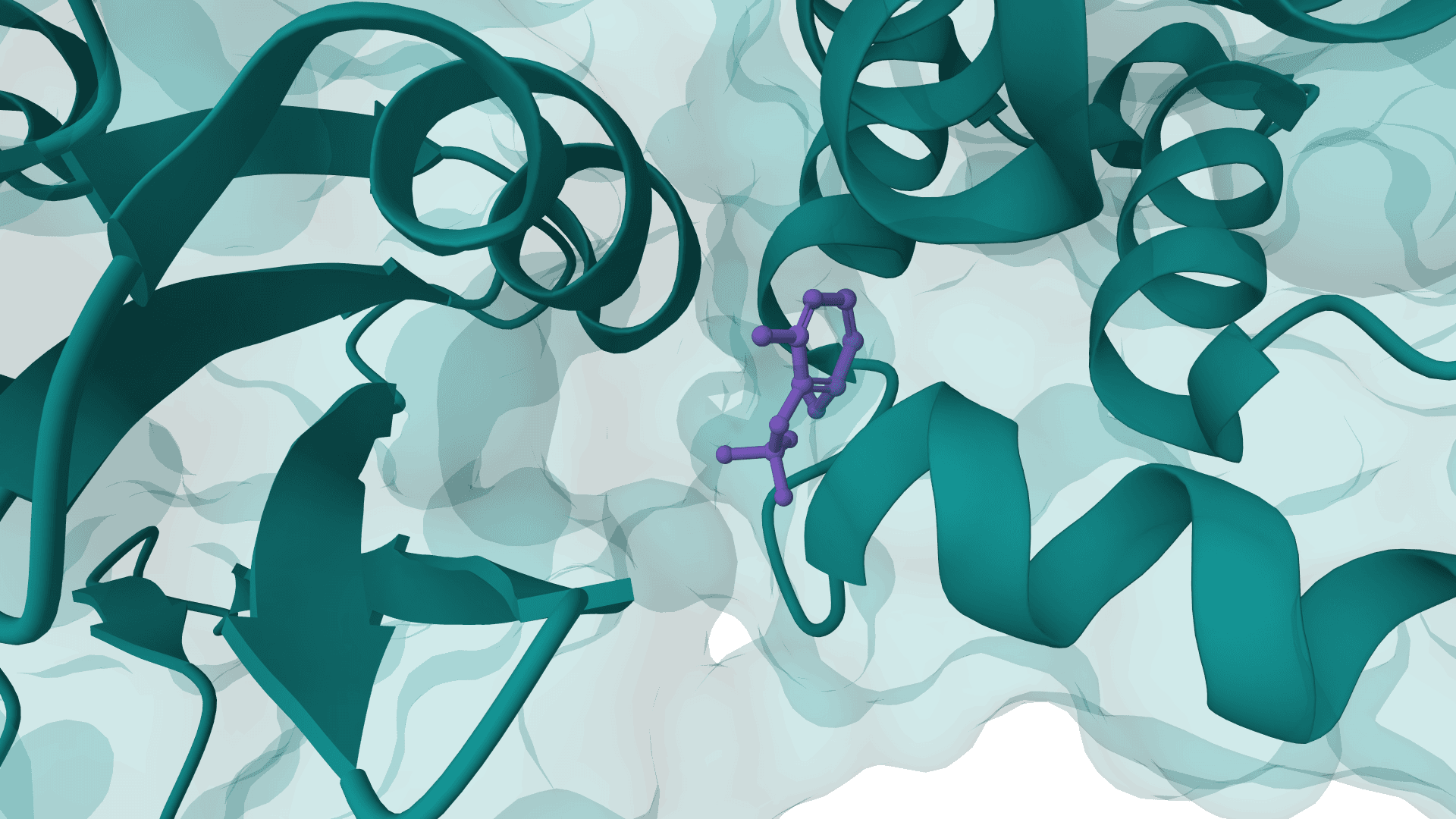
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
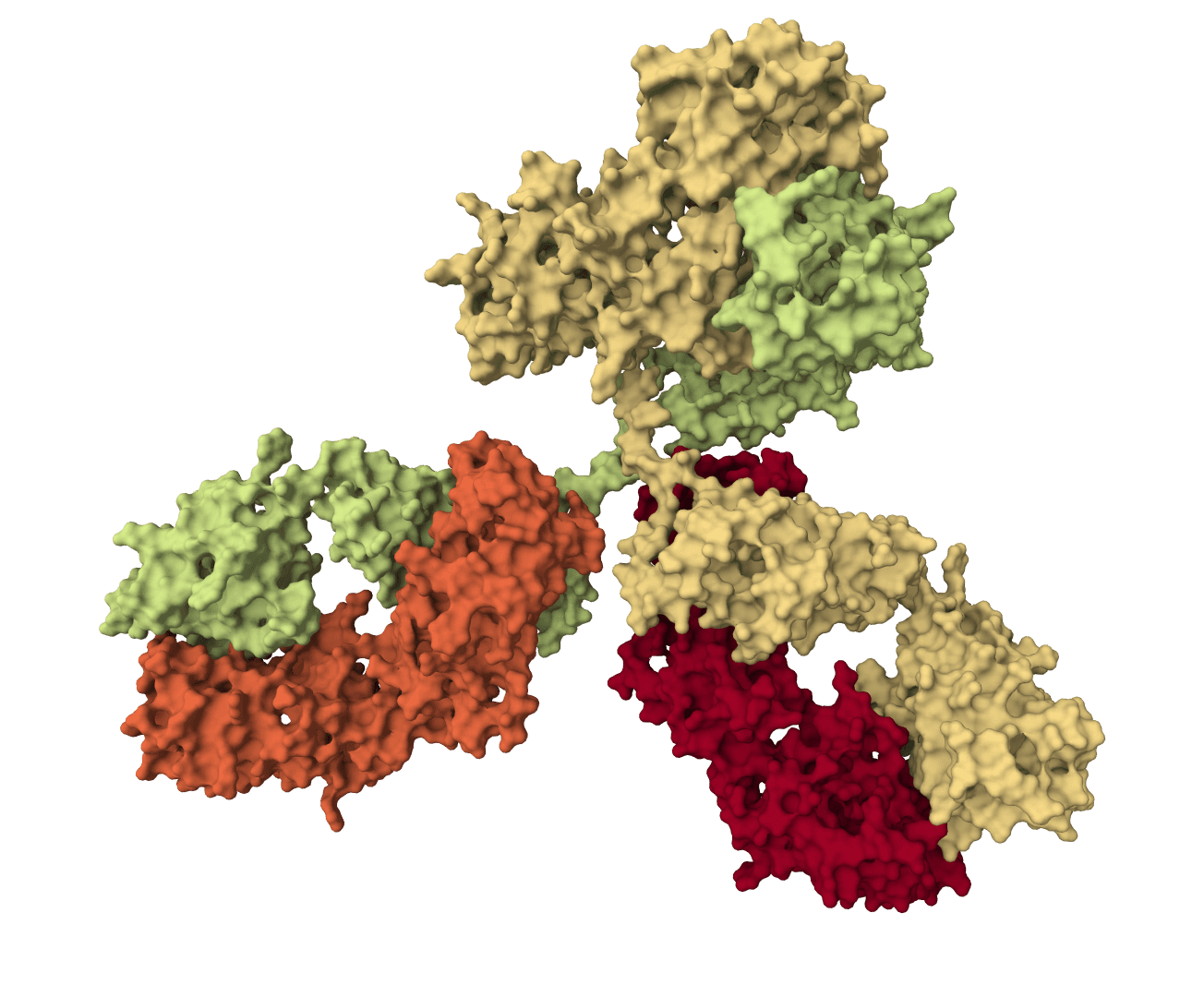
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
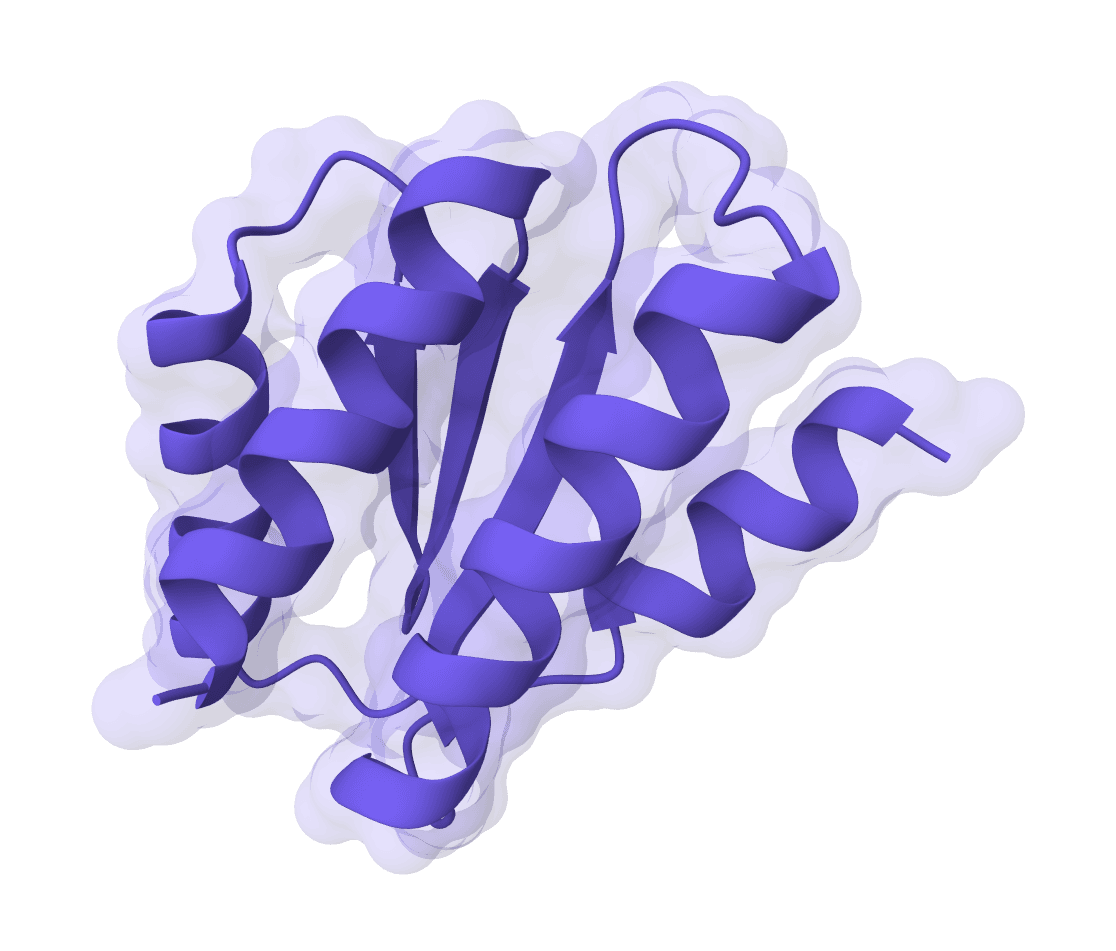
RFdiffusion is a state-of-the-art protein structure generation tool that uses diffusion models to design proteins de novo, create binders, scaffold motifs, and generate symmetric oligomers with atomic precision.
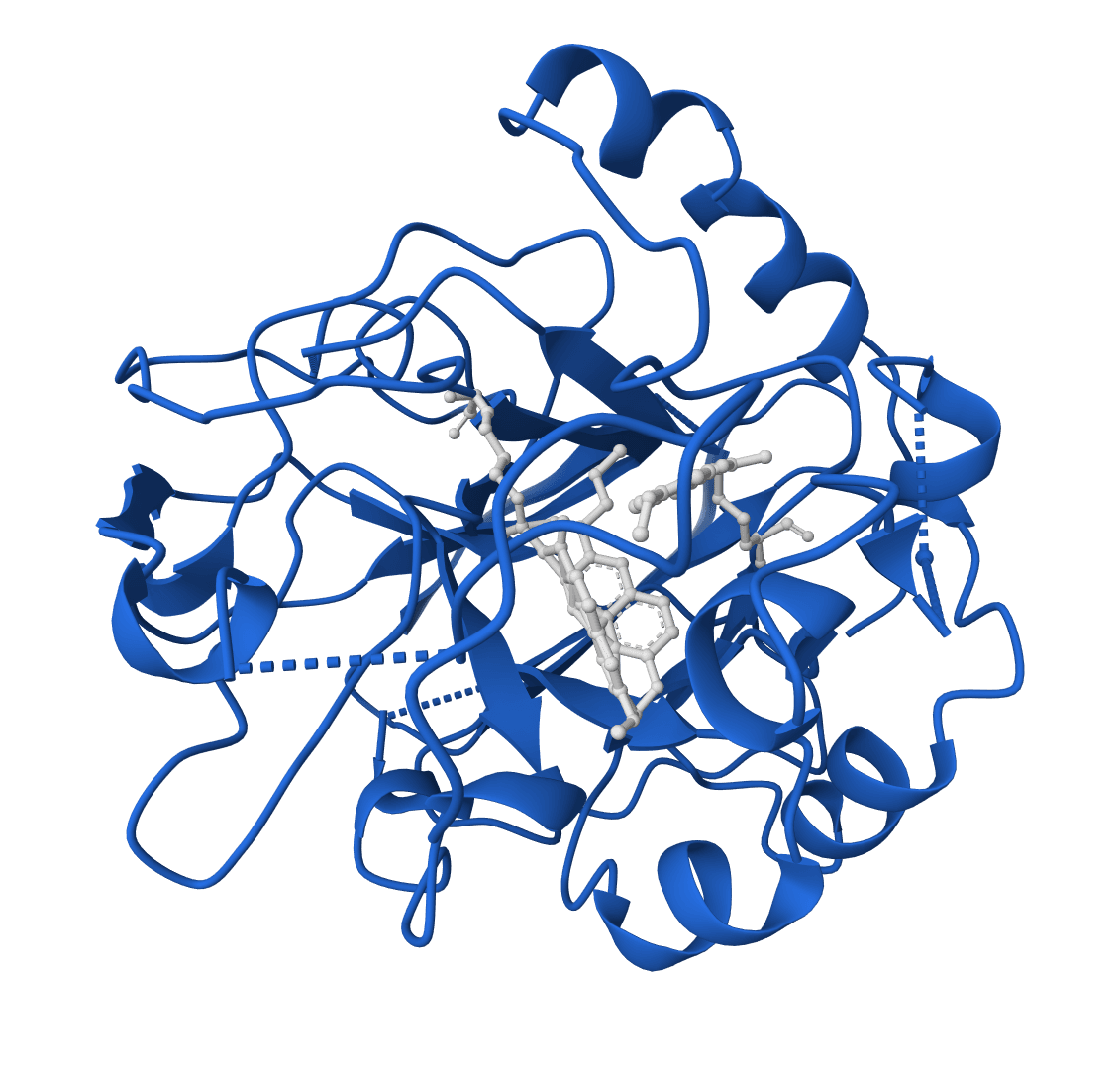
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
AI-powered antibody CDR design using equivariant diffusion models. Generates complementarity-determining region (CDR) sequences and structures for antibody structures and antibody-antigen complexes. Supports single- and multi-CDR co-design, antibody optimization, fixed-backbone sequence design, and structure prediction.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
ProGen2 is Salesforce Research's protein language model suite for prompt-based de novo protein sequence generation and bidirectional sequence likelihood scoring.
ProFam-1 is a protein family language model for family-conditioned sequence generation. Provide a protein family in FASTA, A2M, or A3M format and generate new sequences with model likelihood scores for downstream ranking and screening.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
RFdiffusion is a state-of-the-art protein structure generation tool that uses diffusion models to design proteins de novo, create binders, scaffold motifs, and generate symmetric oligomers with atomic precision.
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
AI-powered antibody CDR design using equivariant diffusion models. Generates complementarity-determining region (CDR) sequences and structures for antibody structures and antibody-antigen complexes. Supports single- and multi-CDR co-design, antibody optimization, fixed-backbone sequence design, and structure prediction.
Configure inputs to begin
Set options on the left, then click “Submit job”.
ODesign is an all-atom generative model for designing protein binders. Built on AlphaFold3-like architecture, it generates binding partners by specifying target epitopes with fine-grained control over interaction sites and binder properties. The model operates at atomic resolution, designing both backbone geometry and sidechain conformations simultaneously.
What distinguishes ODesign from earlier protein design tools is its unified framework for multimodal biomolecular design. While most tools specialize in proteins alone, ODesign handles proteins, nucleic acids, and small molecules within a single generative process, enabling interaction types like protein-binding RNA or DNA-binding ligands that were previously inaccessible.
The system uses diffusion-based generation with conditional control at molecular, motif, and atomic levels. Researchers can design binders for entire protein surfaces or specify precise hotspot residues for targeted interaction design.
ProteinIQ runs ODesign on cloud GPU infrastructure, eliminating the need for local installation or compute resources.
| Input | Description |
|---|---|
Target protein structure | Upload PDB or CIF file, or fetch from RCSB using PDB ID (e.g., 1HSG). Maximum 50 MB. |
| Setting | Description |
|---|---|
Receptor mode | Flexible allows target protein conformational changes during design; Rigid keeps target structure fixed. Flexible mode better captures induced-fit binding but requires more compute. |
Samples per seed | Number of binder designs to generate per random seed (1–10, default 5). Higher values increase diversity but extend runtime. |
Seeds | Random seeds for reproducibility and diversity. Single value (e.g., 42) or comma-separated for multiple runs (e.g., 42,43,44). Each seed produces independent designs. |
Hotspot residues | Target residues for binder interaction (format: A30,A33,A34 or A50-64 for ranges). Leave blank to design binders for the entire target surface. Multiple chains supported (e.g., A100,B50-60). |
Binder length minimum | Minimum binder length in residues (20–200, default 50). |
Binder length maximum | Maximum binder length in residues (50–300, default 100). ODesign samples lengths within this range during generation. |
ODesign generates PDB structures containing the designed binder bound to the target protein. Each output includes:
Multiple structures are returned when using multiple seeds or samples per seed. Download all structures as a ZIP archive or view individual structures in the 3D viewer.
ODesign uses a diffusion-based generative process that progressively denoises random atomic coordinates into structured protein-protein complexes. The model is trained on AlphaFold3-like structure prediction, learning to generate binding partners conditioned on target structures.
Unlike backbone-only methods such as RFdiffusion that generate C-alpha traces requiring separate sidechain packing, ODesign generates full atomic detail in a single pass. This all-atom approach allows the model to account for sidechain interactions during generation rather than post-processing them.
The task-oriented masking mechanism enables control at three levels:
Flexible receptor mode co-generates target-binder interfaces, allowing backbone adjustments in the target protein to accommodate the designed binder. Rigid mode fixes the target structure, generating binders for the provided conformation.
Therapeutic antibody design: Generate novel antibody scaffolds targeting specific epitopes on disease-relevant proteins. Hotspot specification enables focused design for neutralizing epitopes or allosteric sites.
Enzyme inhibitor design: Design protein-based inhibitors for enzyme active sites by specifying catalytic residues as hotspots. Rigid receptor mode maintains catalytic geometry while flexible mode explores induced-fit inhibition.
Protein-protein interaction modulators: Create binders that stabilize or disrupt native protein complexes. Multiple seed generation produces diverse scaffolds for experimental screening.
Nucleic acid-binding protein design: Design proteins that bind DNA or RNA at specified sequences—capabilities not available in protein-only design tools. Useful for genome editing tools and transcription factor engineering.
ODesign outputs are ranked by model confidence but should be evaluated experimentally. Key validation steps:
Not all designs will bind experimentally. Generate multiple seeds and samples to produce diverse candidates for screening. Designs with high model confidence and good structural metrics have higher experimental success rates.
Computational cost: All-atom generation is slower than backbone-only methods. Expect 20–30 minutes for typical runs with multiple seeds and samples.
No binding affinity prediction: ODesign generates structures but does not predict binding affinity (KD or ΔG). Use docking tools like AutoDock Vina or molecular dynamics simulations for affinity estimation.
Validation required: Generated designs are predictions requiring experimental validation. Model confidence does not guarantee experimental binding.
Limited post-translational modifications: The model does not account for glycosylation, phosphorylation, or other modifications that may affect binding.
Training data bias: Performance may degrade for target proteins with unusual folds or non-canonical interactions underrepresented in training data.