All-atom generative diffusion for designing binders, enzymes, and symmetric protein assemblies Learn more
All-atom generative diffusion for designing binders, enzymes, and symmetric protein assemblies Learn more
All-atom generative diffusion for designing binders, enzymes, and symmetric protein assemblies Learn more
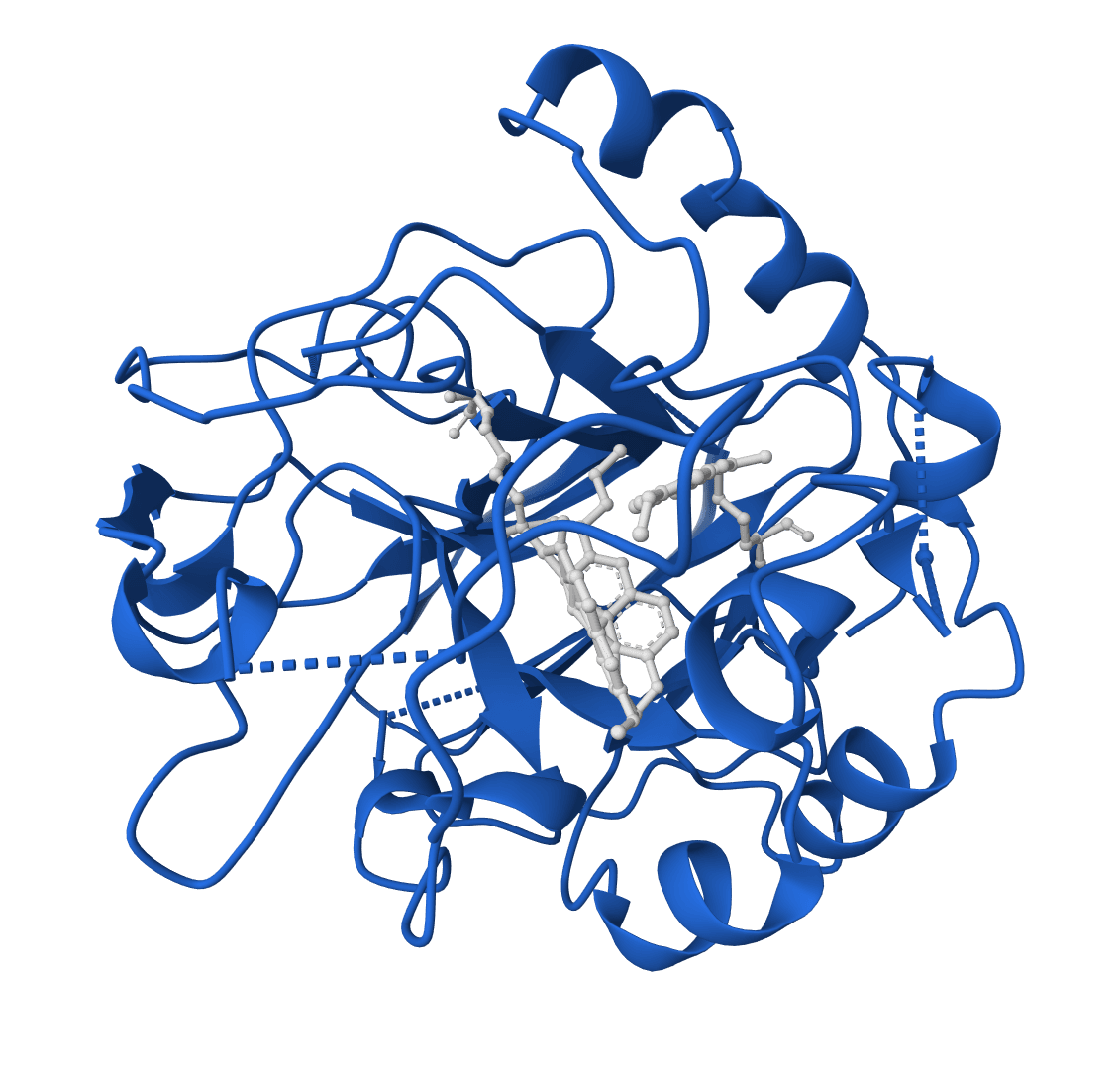
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
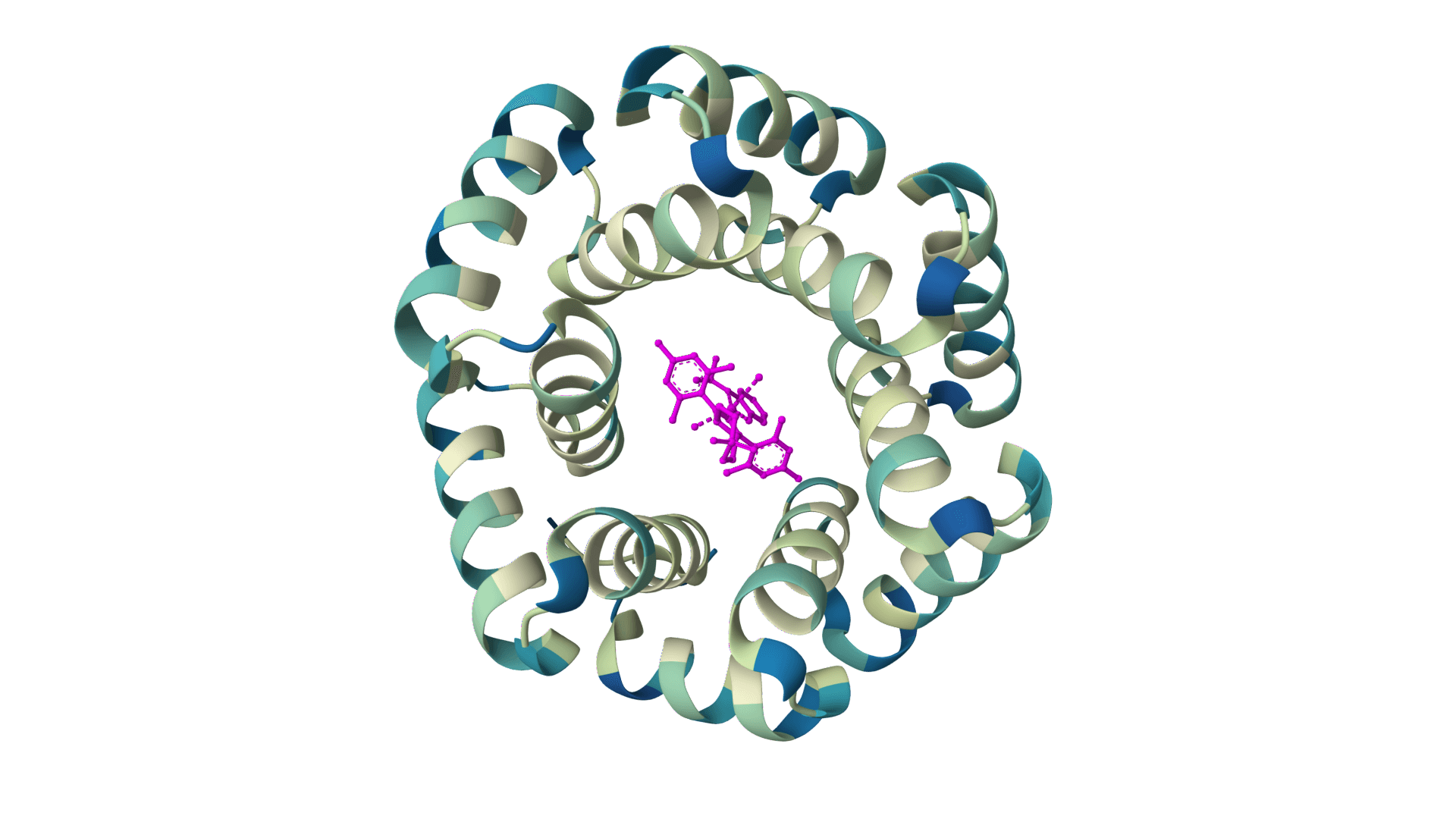
Design protein sequences with atomic context from ligands, metals, and nucleotides. Achieves 63.3% sequence recovery at binding sites, significantly outperforming ProteinMPNN (50.5%).
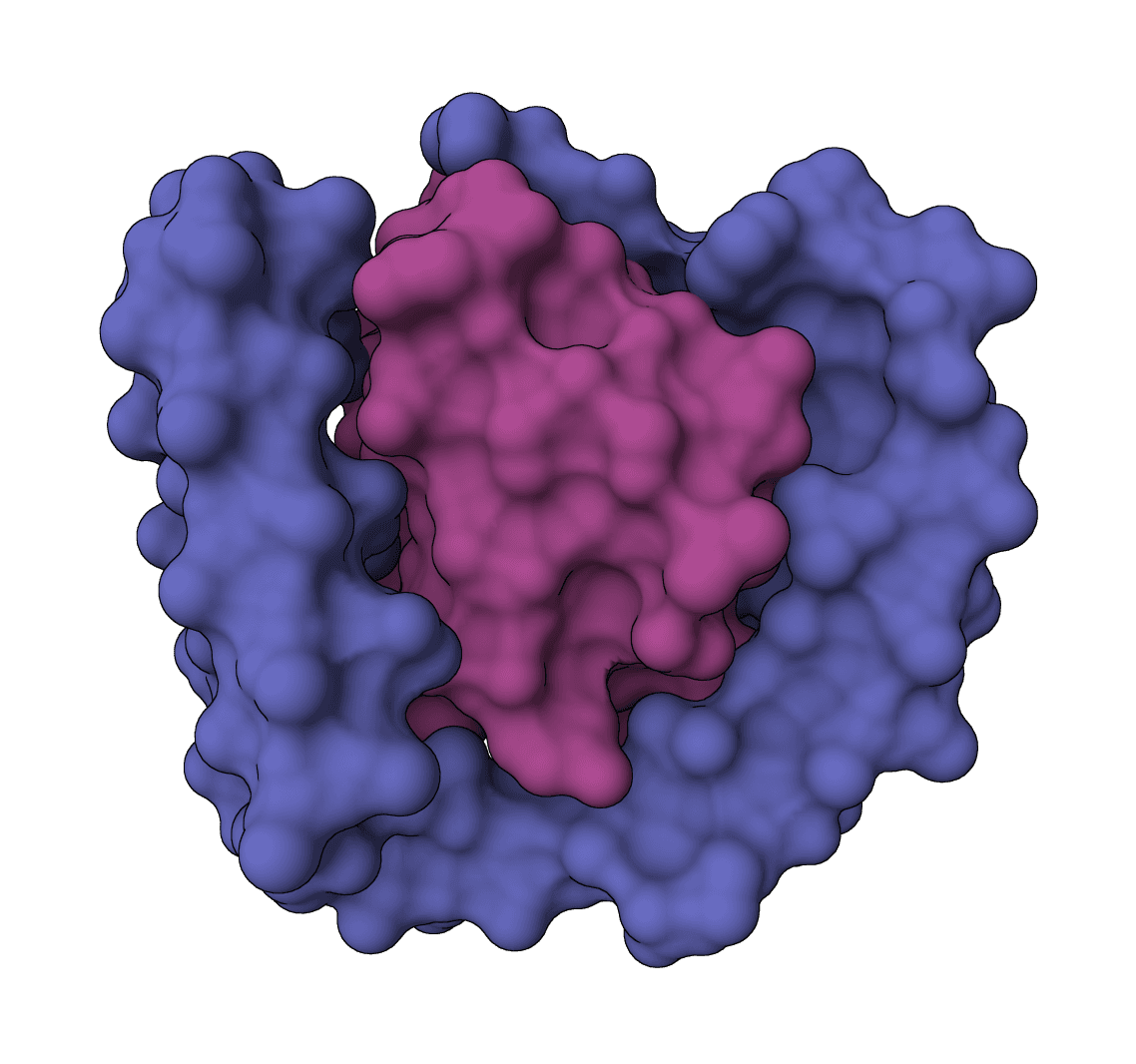
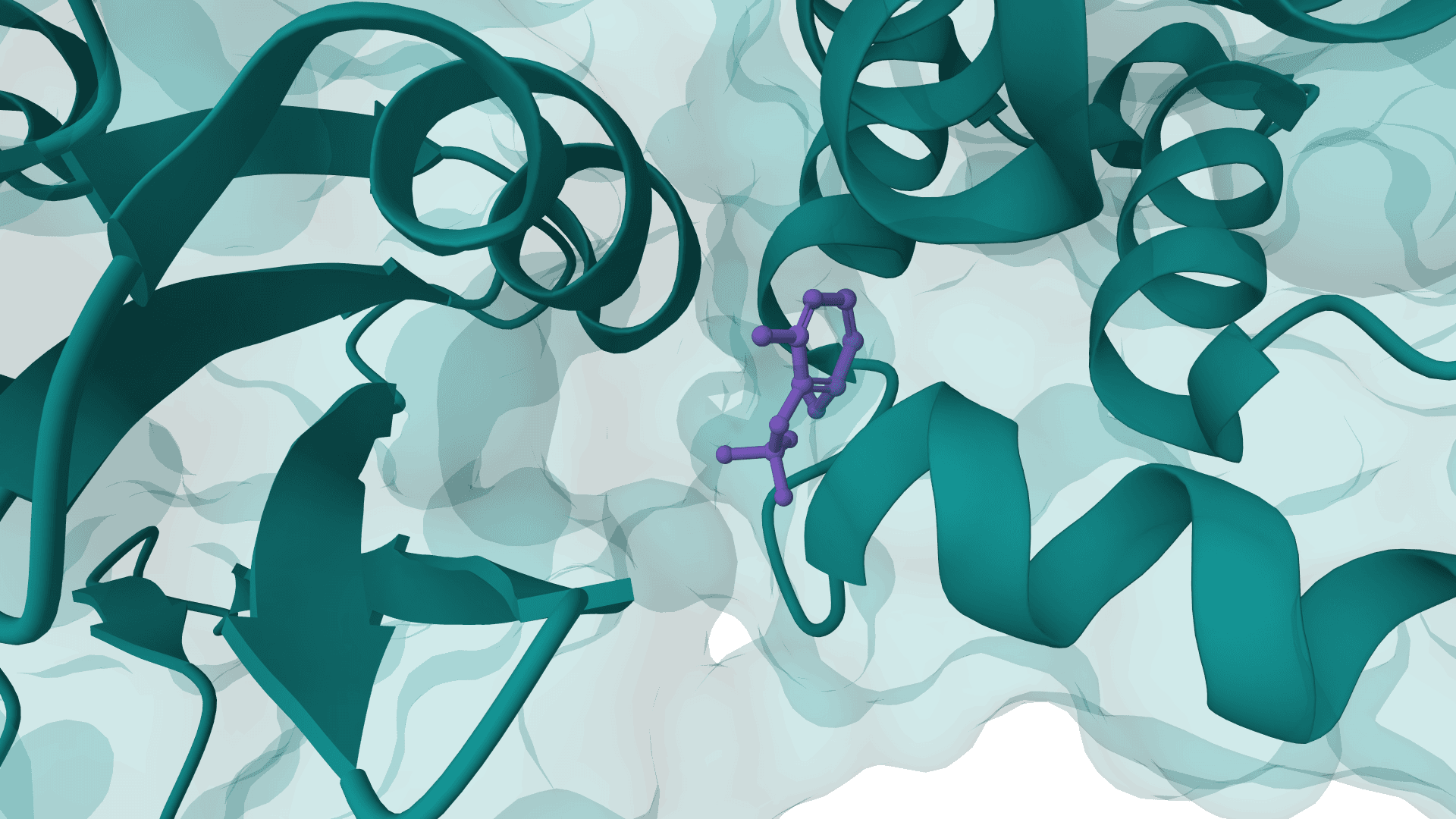
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
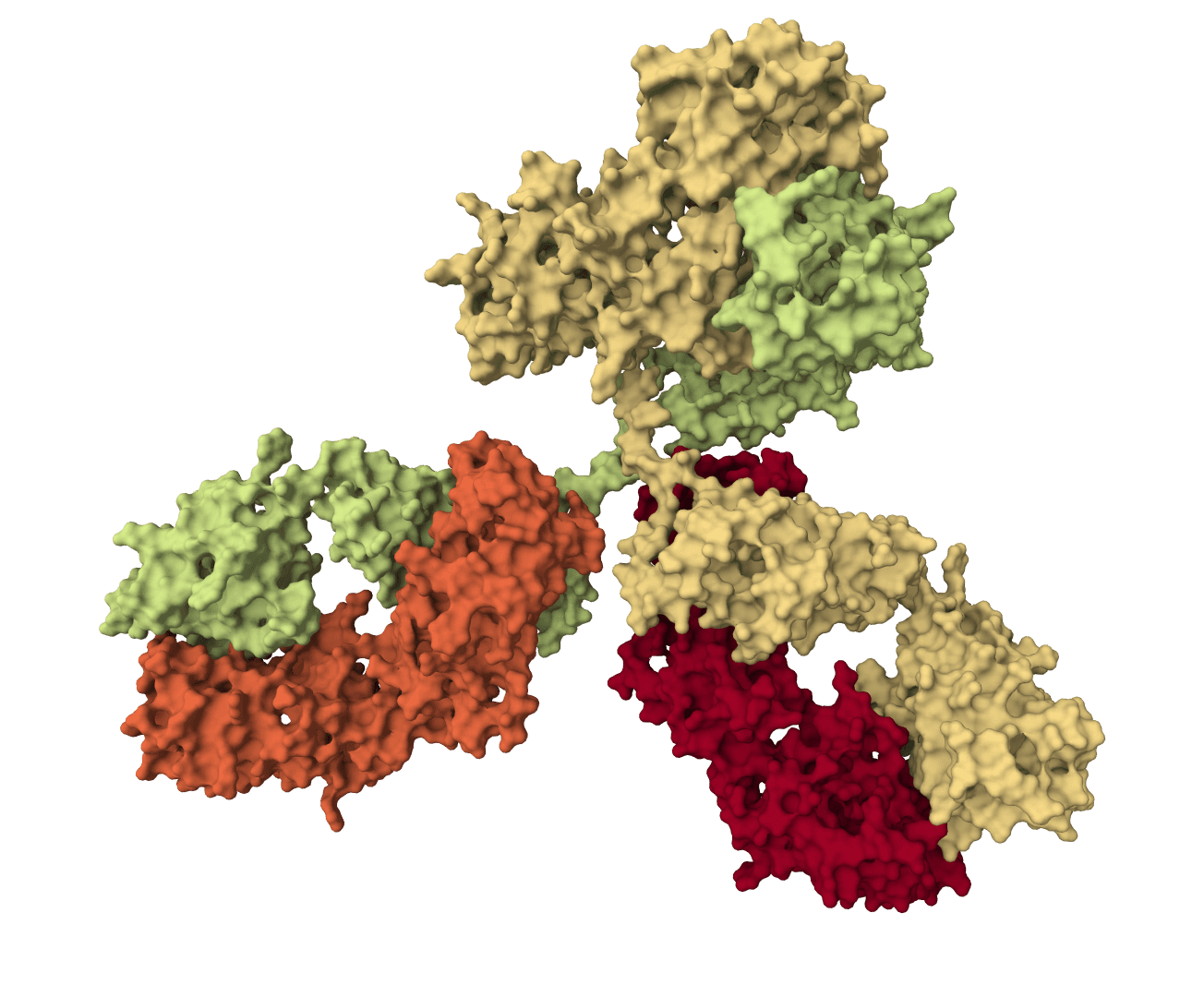
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
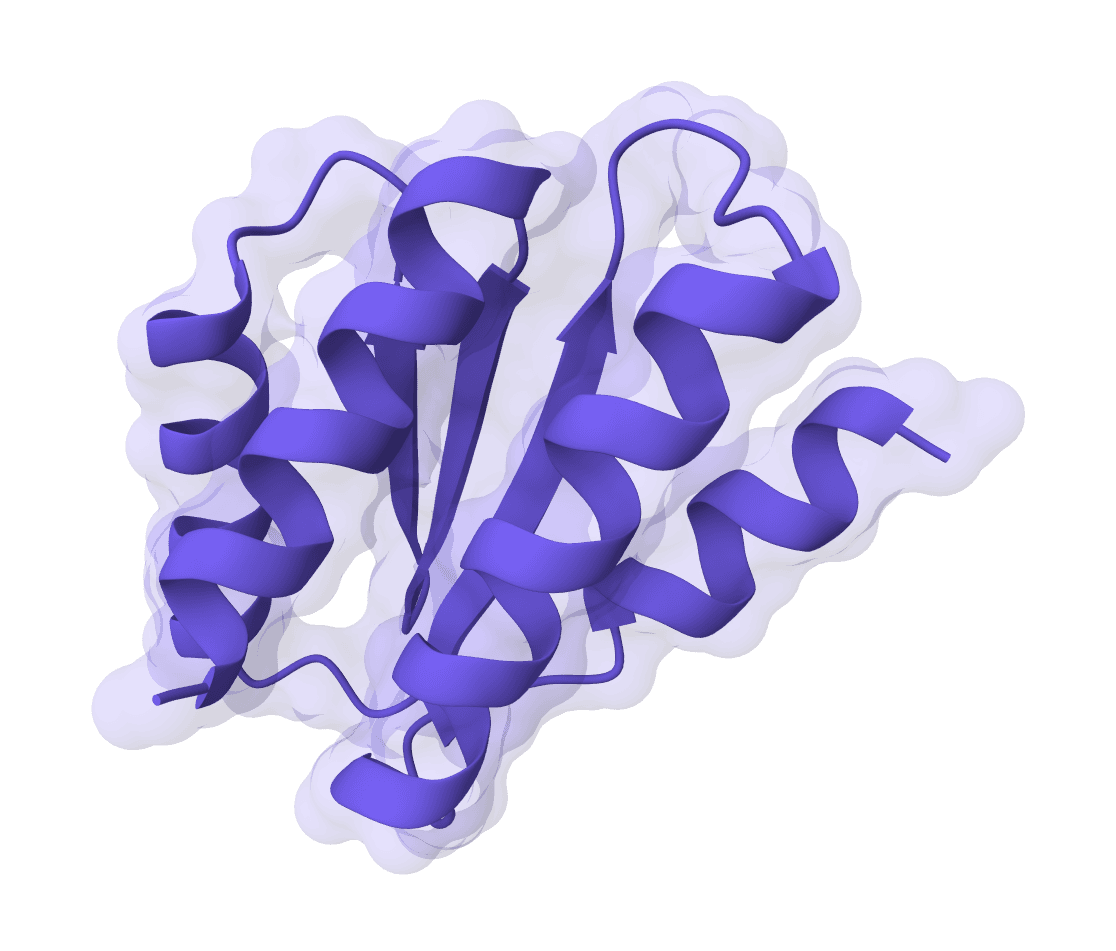
RFdiffusion is a state-of-the-art protein structure generation tool that uses diffusion models to design proteins de novo, create binders, scaffold motifs, and generate symmetric oligomers with atomic precision.
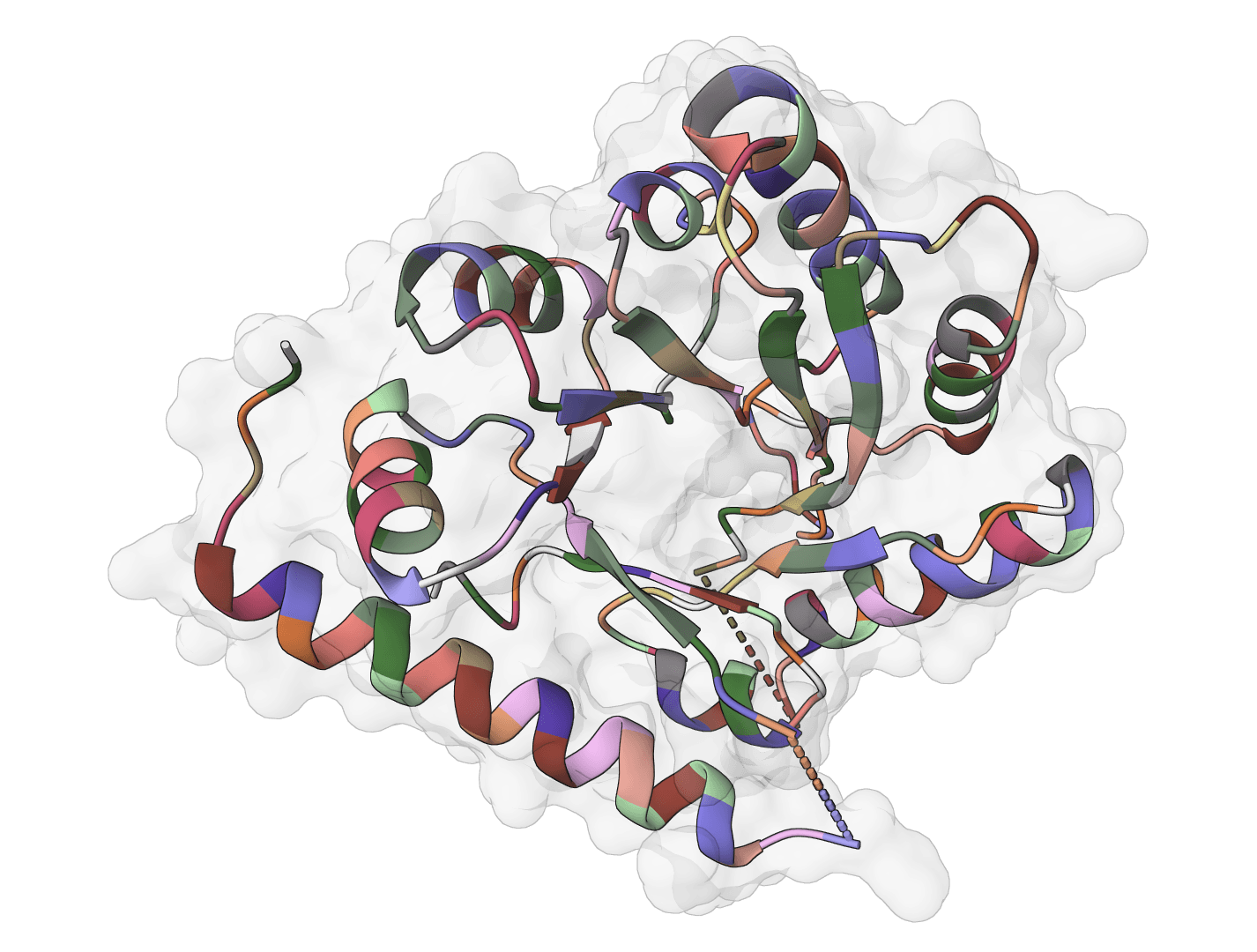
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
Design protein sequences with atomic context from ligands, metals, and nucleotides. Achieves 63.3% sequence recovery at binding sites, significantly outperforming ProteinMPNN (50.5%).
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
RFdiffusion is a state-of-the-art protein structure generation tool that uses diffusion models to design proteins de novo, create binders, scaffold motifs, and generate symmetric oligomers with atomic precision.
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
Configure inputs to begin
Set options on the left, then click “Submit job”.
RFdiffusion3 (RFD3) is a generative diffusion model for de novo protein design. Unlike structure prediction tools that fold existing sequences, RFD3 creates entirely new protein backbones through an iterative denoising process. The model can generate proteins from scratch or design new chains that interact with specified targets.
RFD3 extends the original RFdiffusion architecture with all-atom capabilities, enabling design tasks involving small molecules, metals, and other non-protein components. The model learns to reverse a diffusion process that gradually adds noise to protein structures, generating novel backbones that satisfy specified constraints.
For sequence design after generating backbones, pair RFD3 with LigandMPNN to optimize amino acid sequences. To validate designed structures, use RosettaFold3 or Boltz-2 for structure prediction.
RFD3 operates on a diffusion framework where protein structures are progressively corrupted with Gaussian noise during training. At inference time, the model reverses this process:
The number of diffusion steps controls the quality-speed tradeoff. More steps (100-200) produce higher quality structures but take longer, while fewer steps (10-50) enable rapid prototyping.
RFD3 can incorporate various constraints during generation:
Structural constraints: Fix specific residue positions from a template structure while designing new regions around them. The contig specification language allows precise control over which regions are fixed versus designed.
Hotspot targeting: For binder design, specify which target residue atoms should be contacted by the designed protein. The model orients the new chain to maximize interactions with these hotspots.
Length constraints: Control the size of designed proteins or protein regions using length ranges. The model samples within the specified range during generation.
Provide a target protein structure for constrained design tasks like binder design. Upload a PDB/CIF file or fetch directly from RCSB using a PDB ID (e.g., 4ZXB). The target structure defines the binding interface for binder design or provides structural motifs for scaffolding tasks.
For unconditional generation (creating proteins from scratch), leave this empty and RFD3 will generate novel folds based solely on the specified length.
Select the type of protein design:
Generate multiple independent designs (1-50) in a single job. Each design samples a different trajectory through the diffusion process, producing structural diversity. More designs increase the chance of finding high-quality candidates but require more computation time.
Controls the number of denoising iterations (10-200). Higher values produce more refined structures:
| Steps | Use Case |
|---|---|
| 10-20 | Quick prototyping, initial exploration |
| 50 | Standard quality (default) |
| 100-200 | High-quality final designs |
Specify the length of designed protein regions as a range (e.g., 50-100). The model samples within this range during generation. For binder design, this controls the size of the designed binder chain.
The contig specification provides fine-grained control over the design:
If your uploaded structure has missing residues or gaps, leave Custom contig blank unless you explicitly split the fixed residue ranges to match residues that actually exist in the structure.
Contigs are written as comma-separated pieces:
A1-95 or B5-12 references residues from the uploaded structure50 or 50-100 asks RFD3 to design a region of that length/0 creates a chain breakExamples:
50-100,/0,A1-95 - Design a 50-100 residue binder and place it on a separate chain from target residues A1-A95A40-60,70,A120-170,/0,B3-45,60-80 - Keep residues A40-A60, design a 70-residue segment, keep A120-A170, then start a new chain with B3-B45 followed by a designed 60-80 residue segment100 - Generate a 100-residue protein unconditionallyWhitespace around commas is tolerated by the ProteinIQ form and will be normalized before submission, but the underlying grammar is still comma-separated pieces. Use /0, not a bare /, for chain breaks.
Target specific atoms on the target protein for binder design. This focuses the designed interface on functionally important residues.
Format: JSON object mapping residue IDs to atom names
{
"A64": "CD2,CZ",
"A88": "CG,CZ",
"A96": "CD1,CZ"
}Residue ID format: ChainResidueNumber (e.g., A64 = chain A, residue 64)
The field accepts a JSON object only. Residue IDs must match residues that are actually present in the uploaded target structure.
Common atom names:
N, CA, C, OCD1, CD2, CE1, CE2, CZCB, CG, CDNZ (Lys), OD1/OD2 (Asp), OE1/OE2 (Glu)Hotspot selection dramatically improves binder design success rates by ensuring the designed protein contacts critical interface residues.
Controls how the designed chain is positioned relative to the target:
com): Orient based on the target's center of mass. Use when no specific hotspots are known.Each design generates a PDB file containing the designed backbone coordinates. Files are named sequentially (e.g., design_0.pdb, design_1.pdb). The structures contain:
RFD3 outputs are backbone-only structures requiring sequence design:
While RFD3 doesn't provide explicit confidence scores like folding tools, design quality can be assessed by:
RFD3 excels at: