ColabDock is a protein-protein docking framework that integrates AlphaFold2 with experimental restraints to predict how proteins bind to each other. Developed by Feng and colleagues at Peking University, it was published in Nature Machine Intelligence in August 2024.
Unlike traditional docking methods that use Fast Fourier Transform (FFT) algorithms like ZDOCK, HADDOCK, or ClusPro, ColabDock uses gradient backpropagation to optimize docking poses. This approach automatically integrates the AlphaFold2 energy function with experimental data without requiring retraining.
ColabDock is particularly useful when you have experimental data about protein-protein interfaces—such as cross-linking mass spectrometry (XL-MS), NMR chemical shift perturbation, or covalent labeling experiments. The method outperforms HADDOCK and ClusPro on benchmarks using both simulated and real experimental restraints.
ColabDock operates in two stages. The generation stage uses ColabDesign, a protein design framework built on AlphaFold2, to create initial complex structures.
During generation, the model optimizes sequence representations in logit space while minimizing four loss functions: a monomer distogram loss (preserving individual chain conformations), a restraint loss (bringing specified residues close together), and pLDDT/ipAE losses (ensuring high-quality predictions). The weighted combination of these losses guides the model toward structures that satisfy both the input templates and experimental constraints.
After generation, AlphaFold2 predicts the final complex structure using the generated conformation as a guide. This two-stage approach combines the flexibility of gradient-based optimization with AlphaFold2's accuracy.
ColabDock ranks output structures using a RankingSVM model trained on five features: ipTM (interface quality), contact number, contact pLDDT, number of satisfied restraints, and average error. This ranking helps identify the most likely binding mode when multiple conformations are generated.
ColabDock requires 2-4 protein chain structures as input. You can upload single-chain PDB files directly or fetch chain-specific structures from the RCSB database using identifiers such as 6W63:A.
Each primary protein card should represent one docking partner chain. For typical binary complexes, provide the two interacting chains. For larger assemblies, you can include up to four chains total, but each input must still be a single chain.
If you start from a multichain experimental structure, split it before docking or fetch the exact chain you need. For example, use 6W63:A and 5LU7:B as separate docking inputs instead of uploading the full 5LU7 tetramer as one partner.
If you have a known complex structure, you can upload it as an optional reference/native complex. ProteinIQ uses the extract_rest.py helper from ColabDock to sample restraints from that structure, and the same reference can also be passed through as the native complex for optional RMSD reporting in the final ranking summary.
This is useful when you want faithful native restraint sampling from a known answer or when benchmarking docking poses against an experimentally determined complex.
Unlike the primary docking inputs, the optional reference complex can remain multichain. The important constraint is compatibility: the chain and residue numbering must already match the uploaded docking partners. ProteinIQ normalizes the first matching chains to A,B,C,... for native ColabDock, but it does not perform homolog mapping or residue renumbering.
Experimental restraints encode spatial relationships between residues—typically from cross-linking, NMR, or mutagenesis experiments. ColabDock supports four restraint formats with increasing flexibility.
The simplest restraint type: two specific residues should be in contact. Format each restraint as chain:residue,chain:residue on separate lines.
Example: A:4,B:15 means residue 4 of chain A should contact residue 15 of chain B.
Use 1v1 restraints when you have precise residue-level data, such as identified cross-linked peptide pairs.
When you know one residue contacts somewhere within a region—but not the exact partner—use 1vN restraints. Format: chain:residue,chain:start-end or chain:residue,chain:residue,residue,....
Examples: A:4,B:13-18 means residue 4 of chain A contacts at least one residue between 13-18 of chain B. A:10,B:20,22,24-26 means residue 10 of chain A can satisfy the restraint by contacting residue 20, 22, 24, 25, or 26 of chain B.
This accommodates experimental uncertainty or lower-resolution data like NMR chemical shift perturbation that identifies affected regions rather than specific residues.
For ambiguous data where several restraints could be satisfied, MvN allows you to specify that only some must be true. Format: pair1;pair2;...|min_count.
Example: A:4,B:13-18;A:6,B:13,15,17|1 means at least one of the two candidate 1vN constraints must be satisfied.
This is valuable for noisy experimental data where some cross-links may be false positives.
Some experiments reveal which regions do NOT interact. Repulsive restraints force specified residue pairs to remain distant. Format: chain:residue,chain:residue.
Example: A:6,B:18 means residue 6 of chain A should be far from residue 18 of chain B.
Use repulsive restraints when you have negative data—regions that definitively don't form the interface.
When using a reference complex, these settings map directly onto the extract_rest.py sampler:
A,B.1v1, 1vN, MvN, or repulsive.--num value.--N value used for 1vN and MvN.--M value used when sampling MvN restraints.8.0 Å default is standard for cross-linking studies and is also passed to native restraint sampling.fixed_chains setting. Use semicolon-separated groups such as A,B;C,D to keep the relative positions within those groups fixed during docking.crop_len setting. This enables segment-based optimization for larger complexes when GPU memory is limiting.native ColabDock writes three artifact folders: gen, pred, and docked. ProteinIQ returns all of them in the Files tab. The docked folder contains the ranked top 5 docked complexes, while summary.txt records the native ipTM score, the number of satisfied restraints, and optional RMSD values when a reference/native complex was provided.
Examine the top 3-5 poses rather than relying solely on rank 1. Multiple similar top poses suggest a confident prediction, while diverse poses may indicate conformational flexibility or an uncertain binding mode.
If you provided restraints, check how many are satisfied in each pose. A good prediction should satisfy most attractive restraints while avoiding contacts flagged as repulsive. ipTM is used as the primary interface-quality score in the ProteinIQ data view, but the raw summary.txt is also available for download.
| Tool | Approach | Restraint support | Best for |
|---|---|---|---|
| ColabDock | AF2 + gradient backpropagation | 1v1, 1vN, MvN, repulsive | Data-driven docking with XL-MS/NMR |
| LightDock | Glowworm Swarm Optimization | Limited | Blind docking, flexibility |
| HADDOCK | FFT + refinement | Ambiguous restraints | NMR-driven docking |
| ClusPro | FFT + clustering | None | Blind docking screening |
ColabDock is the preferred choice when you have experimental restraint data. For blind docking without prior knowledge, consider LightDock or ClusPro.
ColabDock has a maximum restraint distance of 22 Å, determined by AlphaFold2's distance map upper limit. This restricts compatibility to shorter cross-linking reagents; longer-range XL-MS data cannot be directly incorporated.
The method can process complexes up to approximately 1,200 residues on an NVIDIA A100 GPU. Larger assemblies require segment-based optimization or splitting into subcomplexes.
Without experimental restraints, ColabDock may not outperform dedicated blind docking tools. The method's strength lies in integrating experimental data with deep learning predictions.
AF2Dock adapts AlphaFold2-style co-folding for structure-based protein-protein docking. It docks receptor and ligand protein structures with flow-matching refinement and ranks sampled complexes by iPTM.
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
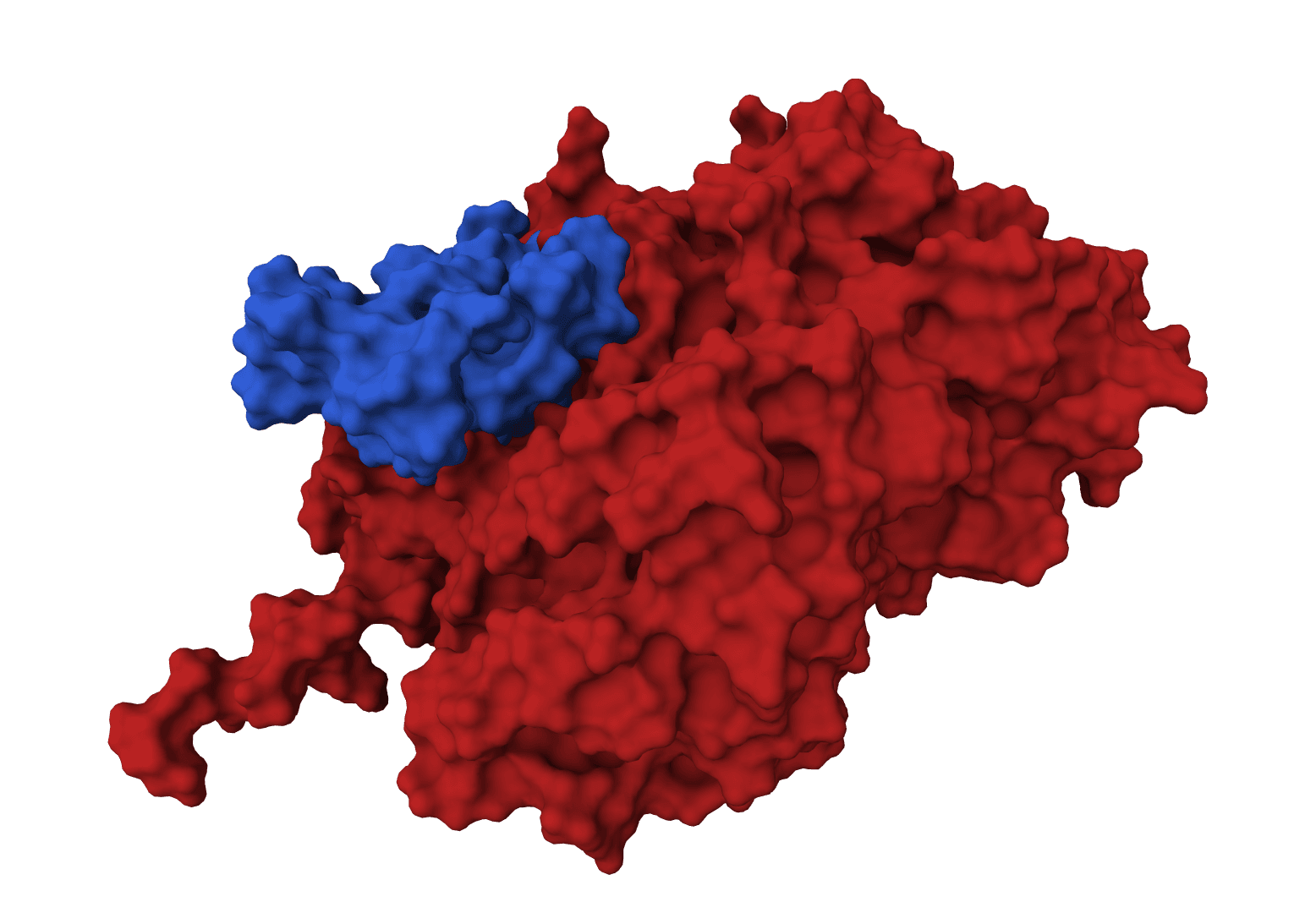
GeoDock predicts flexible protein-protein docking complexes from two separate protein structures using a multi-track iterative transformer and the DIPS 0.3 checkpoint from the Gray Lab release.
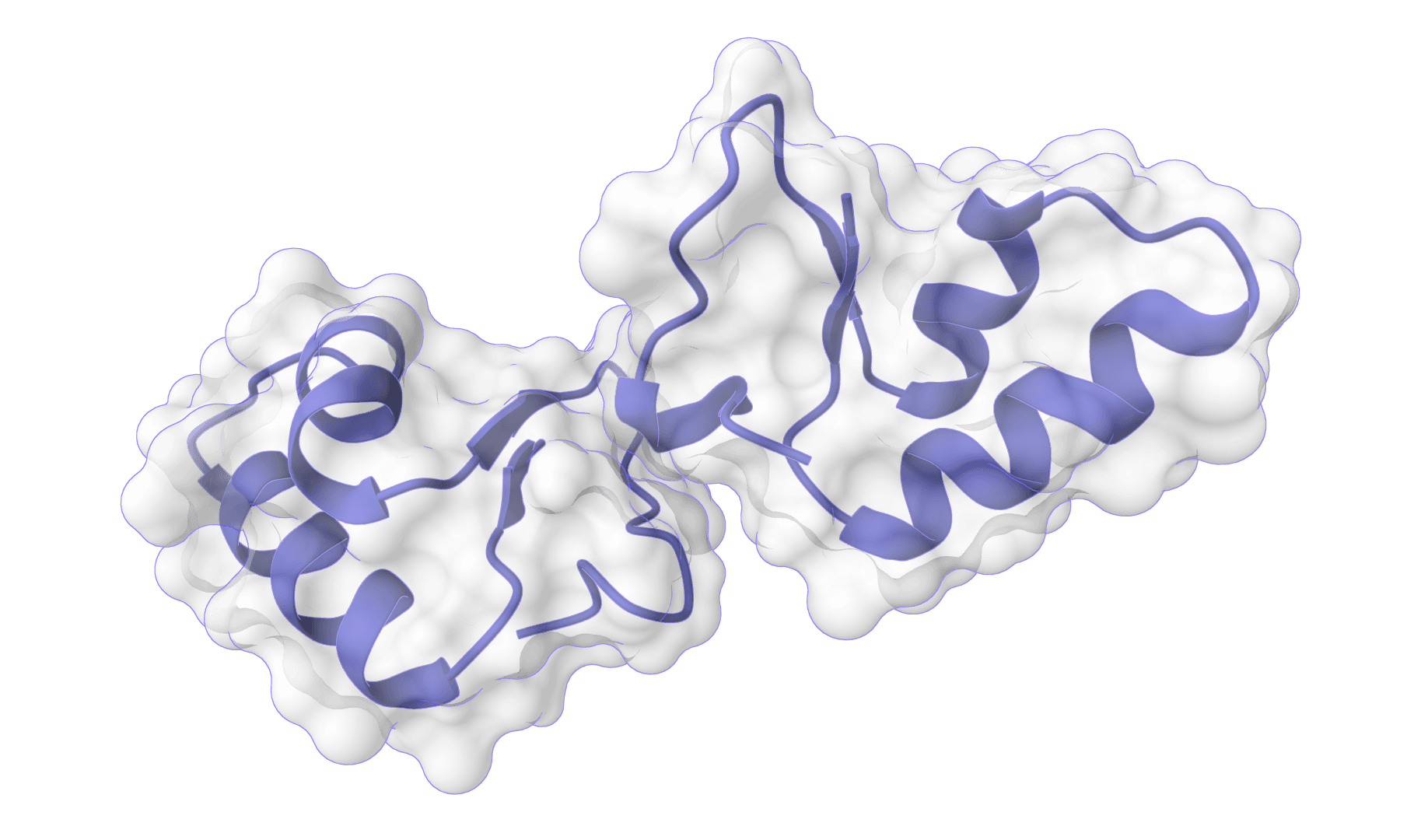
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
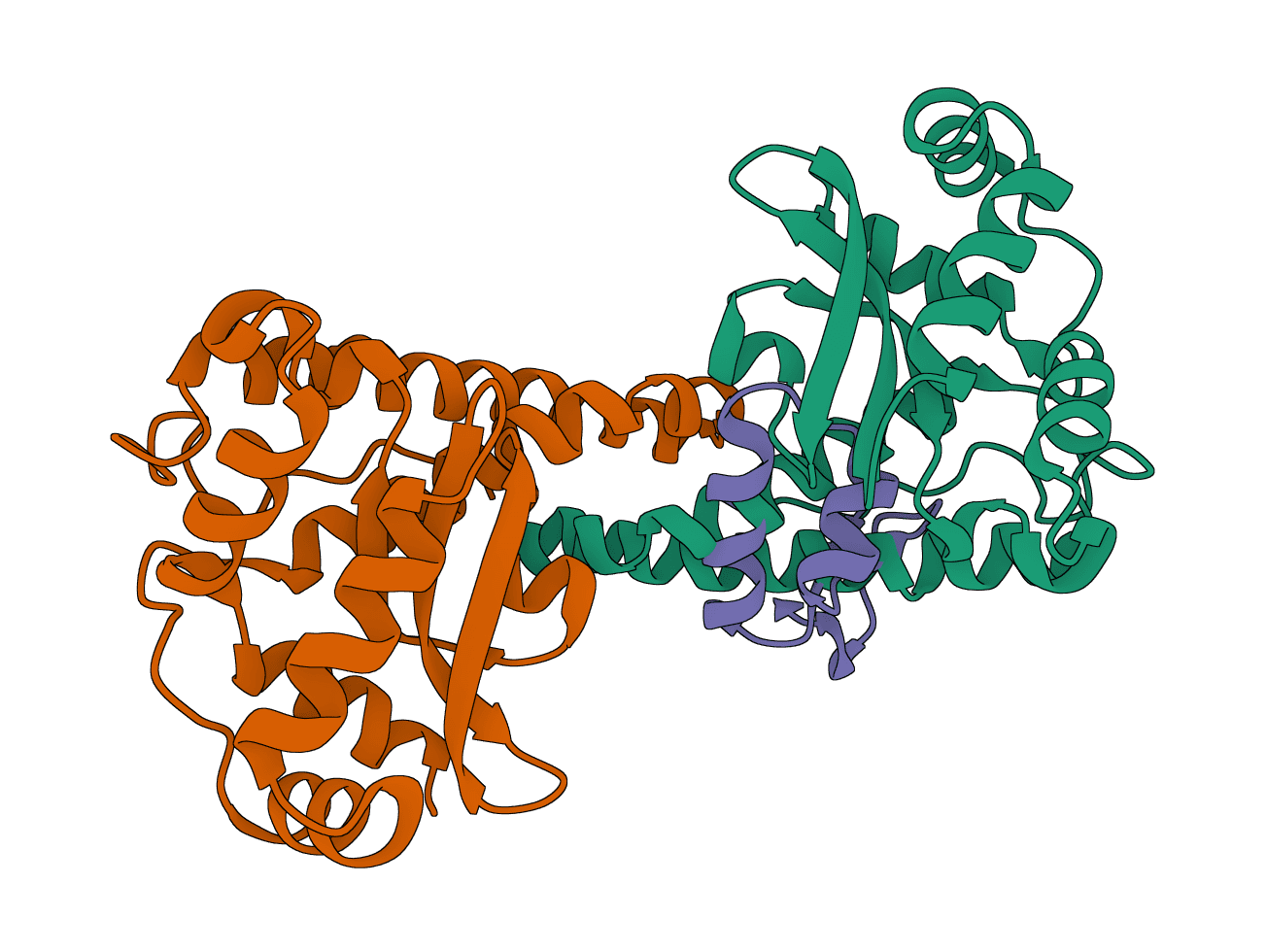
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
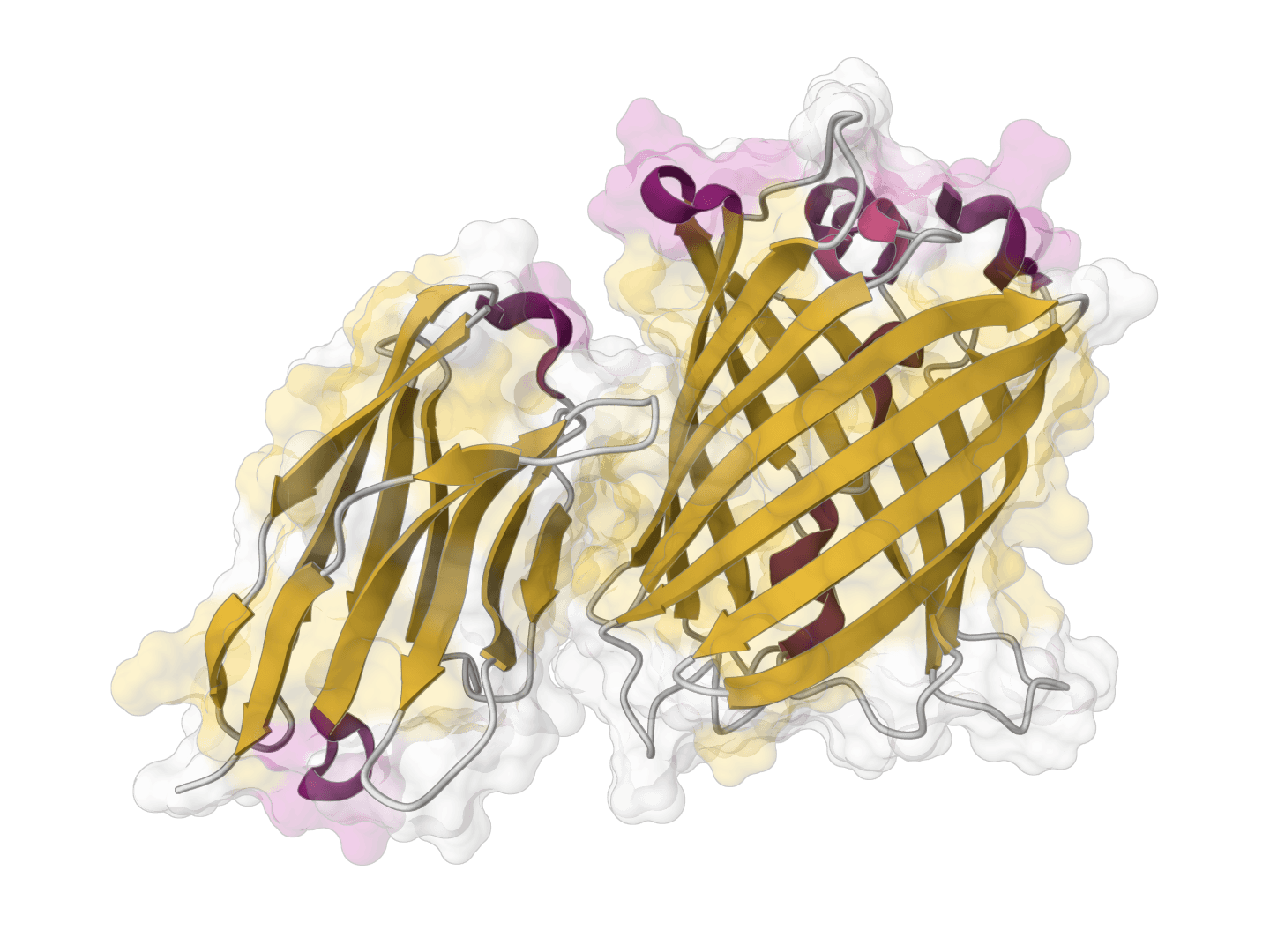
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
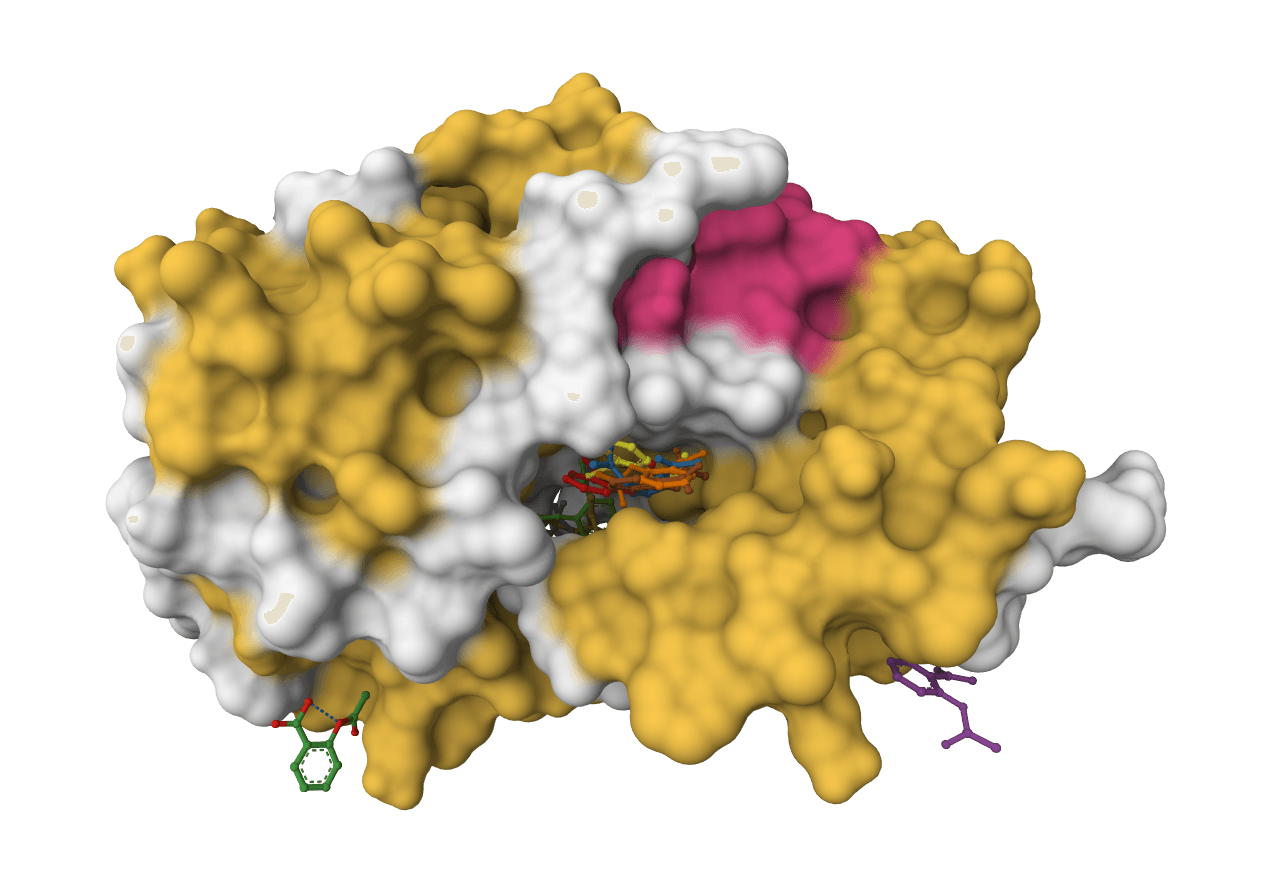
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
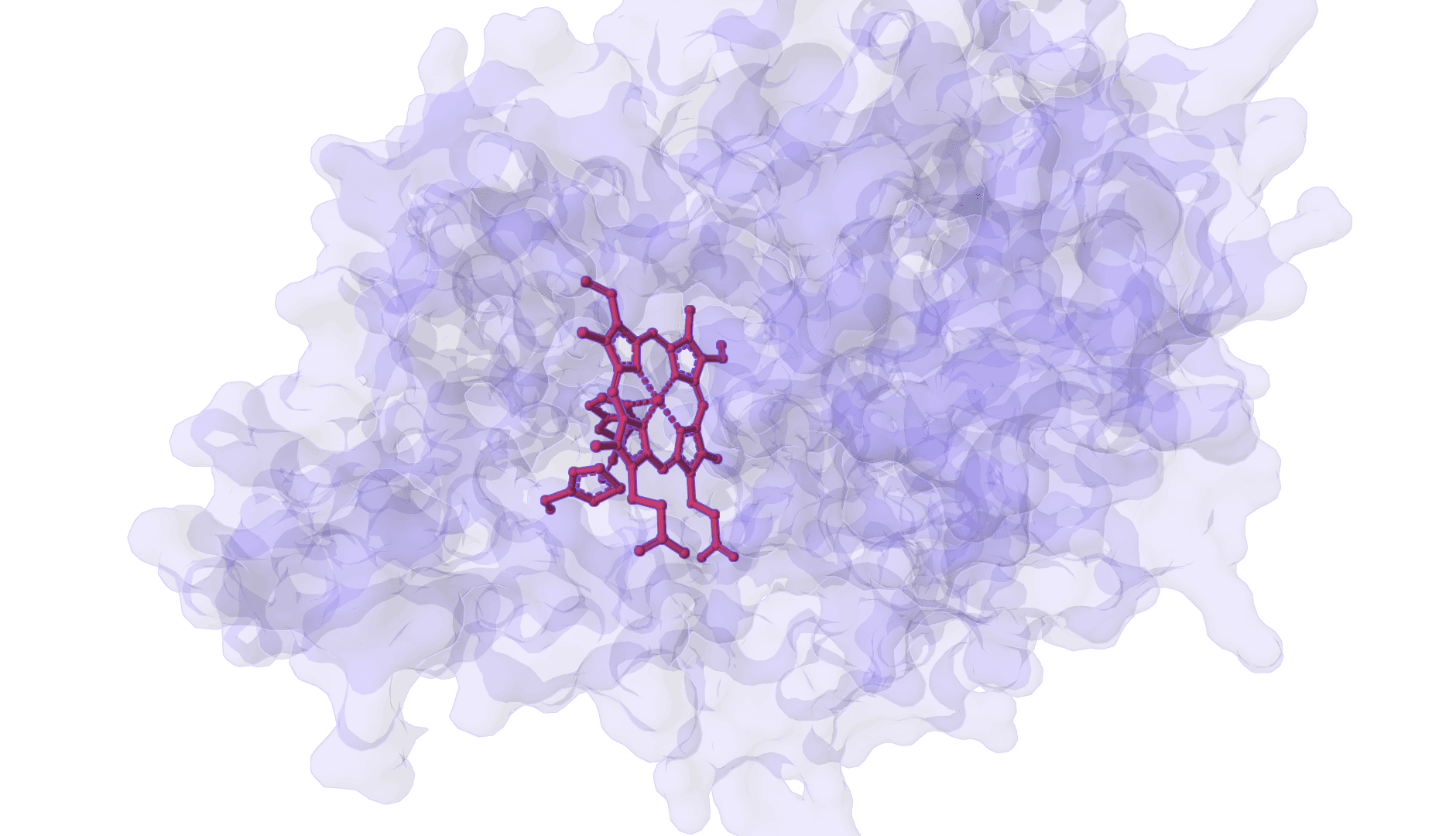
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
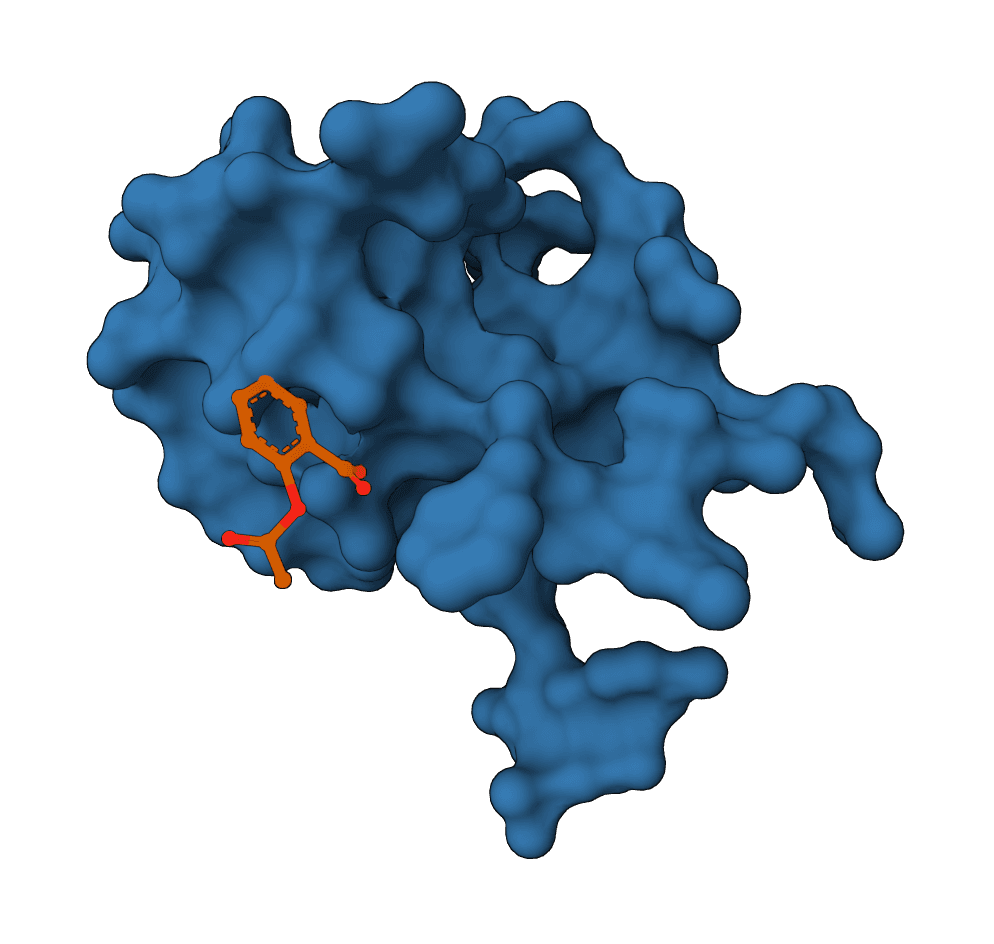
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
ColabDock is a protein-protein docking framework that integrates AlphaFold2 with experimental restraints to predict how proteins bind to each other. Developed by Feng and colleagues at Peking University, it was published in Nature Machine Intelligence in August 2024.
Unlike traditional docking methods that use Fast Fourier Transform (FFT) algorithms like ZDOCK, HADDOCK, or ClusPro, ColabDock uses gradient backpropagation to optimize docking poses. This approach automatically integrates the AlphaFold2 energy function with experimental data without requiring retraining.
ColabDock is particularly useful when you have experimental data about protein-protein interfaces—such as cross-linking mass spectrometry (XL-MS), NMR chemical shift perturbation, or covalent labeling experiments. The method outperforms HADDOCK and ClusPro on benchmarks using both simulated and real experimental restraints.
ColabDock operates in two stages. The generation stage uses ColabDesign, a protein design framework built on AlphaFold2, to create initial complex structures.
During generation, the model optimizes sequence representations in logit space while minimizing four loss functions: a monomer distogram loss (preserving individual chain conformations), a restraint loss (bringing specified residues close together), and pLDDT/ipAE losses (ensuring high-quality predictions). The weighted combination of these losses guides the model toward structures that satisfy both the input templates and experimental constraints.
After generation, AlphaFold2 predicts the final complex structure using the generated conformation as a guide. This two-stage approach combines the flexibility of gradient-based optimization with AlphaFold2's accuracy.
ColabDock ranks output structures using a RankingSVM model trained on five features: ipTM (interface quality), contact number, contact pLDDT, number of satisfied restraints, and average error. This ranking helps identify the most likely binding mode when multiple conformations are generated.
ColabDock requires 2-4 protein chain structures as input. You can upload single-chain PDB files directly or fetch chain-specific structures from the RCSB database using identifiers such as 6W63:A.
Each primary protein card should represent one docking partner chain. For typical binary complexes, provide the two interacting chains. For larger assemblies, you can include up to four chains total, but each input must still be a single chain.
If you start from a multichain experimental structure, split it before docking or fetch the exact chain you need. For example, use 6W63:A and 5LU7:B as separate docking inputs instead of uploading the full 5LU7 tetramer as one partner.
If you have a known complex structure, you can upload it as an optional reference/native complex. ProteinIQ uses the extract_rest.py helper from ColabDock to sample restraints from that structure, and the same reference can also be passed through as the native complex for optional RMSD reporting in the final ranking summary.
This is useful when you want faithful native restraint sampling from a known answer or when benchmarking docking poses against an experimentally determined complex.
Unlike the primary docking inputs, the optional reference complex can remain multichain. The important constraint is compatibility: the chain and residue numbering must already match the uploaded docking partners. ProteinIQ normalizes the first matching chains to A,B,C,... for native ColabDock, but it does not perform homolog mapping or residue renumbering.
Experimental restraints encode spatial relationships between residues—typically from cross-linking, NMR, or mutagenesis experiments. ColabDock supports four restraint formats with increasing flexibility.
The simplest restraint type: two specific residues should be in contact. Format each restraint as chain:residue,chain:residue on separate lines.
Example: A:4,B:15 means residue 4 of chain A should contact residue 15 of chain B.
Use 1v1 restraints when you have precise residue-level data, such as identified cross-linked peptide pairs.
When you know one residue contacts somewhere within a region—but not the exact partner—use 1vN restraints. Format: chain:residue,chain:start-end or chain:residue,chain:residue,residue,....
Examples: A:4,B:13-18 means residue 4 of chain A contacts at least one residue between 13-18 of chain B. A:10,B:20,22,24-26 means residue 10 of chain A can satisfy the restraint by contacting residue 20, 22, 24, 25, or 26 of chain B.
This accommodates experimental uncertainty or lower-resolution data like NMR chemical shift perturbation that identifies affected regions rather than specific residues.
For ambiguous data where several restraints could be satisfied, MvN allows you to specify that only some must be true. Format: pair1;pair2;...|min_count.
Example: A:4,B:13-18;A:6,B:13,15,17|1 means at least one of the two candidate 1vN constraints must be satisfied.
This is valuable for noisy experimental data where some cross-links may be false positives.
Some experiments reveal which regions do NOT interact. Repulsive restraints force specified residue pairs to remain distant. Format: chain:residue,chain:residue.
Example: A:6,B:18 means residue 6 of chain A should be far from residue 18 of chain B.
Use repulsive restraints when you have negative data—regions that definitively don't form the interface.
When using a reference complex, these settings map directly onto the extract_rest.py sampler:
A,B.1v1, 1vN, MvN, or repulsive.--num value.--N value used for 1vN and MvN.--M value used when sampling MvN restraints.8.0 Å default is standard for cross-linking studies and is also passed to native restraint sampling.fixed_chains setting. Use semicolon-separated groups such as A,B;C,D to keep the relative positions within those groups fixed during docking.crop_len setting. This enables segment-based optimization for larger complexes when GPU memory is limiting.native ColabDock writes three artifact folders: gen, pred, and docked. ProteinIQ returns all of them in the Files tab. The docked folder contains the ranked top 5 docked complexes, while summary.txt records the native ipTM score, the number of satisfied restraints, and optional RMSD values when a reference/native complex was provided.
Examine the top 3-5 poses rather than relying solely on rank 1. Multiple similar top poses suggest a confident prediction, while diverse poses may indicate conformational flexibility or an uncertain binding mode.
If you provided restraints, check how many are satisfied in each pose. A good prediction should satisfy most attractive restraints while avoiding contacts flagged as repulsive. ipTM is used as the primary interface-quality score in the ProteinIQ data view, but the raw summary.txt is also available for download.
| Tool | Approach | Restraint support | Best for |
|---|---|---|---|
| ColabDock | AF2 + gradient backpropagation | 1v1, 1vN, MvN, repulsive | Data-driven docking with XL-MS/NMR |
| LightDock | Glowworm Swarm Optimization | Limited | Blind docking, flexibility |
| HADDOCK | FFT + refinement | Ambiguous restraints | NMR-driven docking |
| ClusPro | FFT + clustering | None | Blind docking screening |
ColabDock is the preferred choice when you have experimental restraint data. For blind docking without prior knowledge, consider LightDock or ClusPro.
ColabDock has a maximum restraint distance of 22 Å, determined by AlphaFold2's distance map upper limit. This restricts compatibility to shorter cross-linking reagents; longer-range XL-MS data cannot be directly incorporated.
The method can process complexes up to approximately 1,200 residues on an NVIDIA A100 GPU. Larger assemblies require segment-based optimization or splitting into subcomplexes.
Without experimental restraints, ColabDock may not outperform dedicated blind docking tools. The method's strength lies in integrating experimental data with deep learning predictions.
AF2Dock adapts AlphaFold2-style co-folding for structure-based protein-protein docking. It docks receptor and ligand protein structures with flow-matching refinement and ranks sampled complexes by iPTM.
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
GeoDock predicts flexible protein-protein docking complexes from two separate protein structures using a multi-track iterative transformer and the DIPS 0.3 checkpoint from the Gray Lab release.
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.