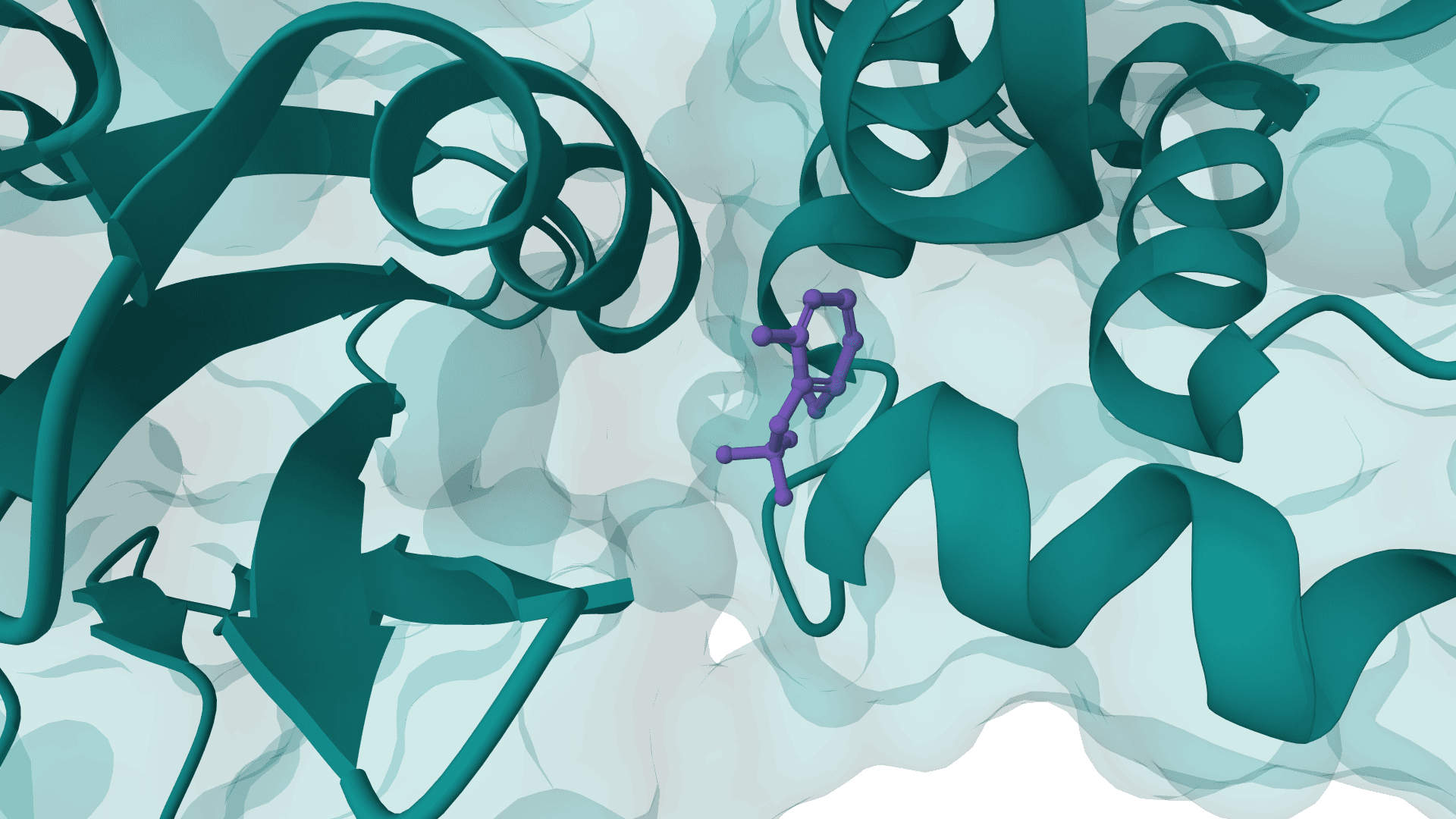
PocketFlow is a structure-based deep generative model that designs novel drug-like molecules inside protein binding pockets. Published in Nature Machine Intelligence in March 2024 by researchers at Sichuan University, it combines autoregressive flow modeling with explicit chemical knowledge to generate molecules with 100% chemical validity.
What sets PocketFlow apart is its experimental validation. The authors applied PocketFlow to design inhibitors for two epigenetic targets (HAT1 and YTHDC1) and successfully obtained wet-lab-validated bioactive lead compounds. This makes PocketFlow the first structure-based molecular deep generative model with experimental validation of designed molecules.
In computational benchmarks on the CrossDocked2020 dataset, PocketFlow outperforms previous methods while maintaining perfect chemical validity and high drug-likeness scores.
PocketFlow generates molecules atom-by-atom within a protein binding pocket using an autoregressive approach. At each step, the model decides what atom type to add, where to place it in 3D space, and how to connect it to existing atoms. Chemical rules guide these decisions to ensure valid molecules.
The core of PocketFlow is the Geometric Double Bottleneck Perceptron (GDBP), an SE(3)-equivariant graph neural network that models the 3D geometry of the protein-ligand complex. GDBP improves upon earlier geometric neural networks (GVP and GBP) by adding bottleneck layers for both scalar and vector features.
This architecture processes 3D coordinates directly while maintaining equivariance to rotations and translations. The model can generate atom positions in 3D space without needing to first predict internal coordinates.
The autoregressive generation uses three specialized components working together:
Atom Flow predicts the type of each new atom (carbon, nitrogen, oxygen, etc.) using a normalizing flow. This probabilistic approach captures the distribution of atom types conditioned on the current molecular state and pocket environment.
Position Predictor determines where to place each new atom in 3D space relative to the binding pocket. The GDBP network encodes spatial relationships between existing atoms, protein residues, and potential placement sites.
Bond Flow predicts connectivity between the new atom and existing atoms using another normalizing flow. This component receives explicit chemical knowledge guidance to ensure reasonable bond patterns.
Unlike purely data-driven approaches, PocketFlow incorporates chemical knowledge directly into the generation process. The bond predictor checks whether proposed bonds satisfy valence rules and reasonable bonding patterns.
If the model proposes an unreasonable bond, it resamples until finding a valid connection. This explicit guidance is critical—ablation studies show that removing chemical constraints significantly degrades both validity and drug-likeness of generated molecules.
PocketFlow uses a two-stage training process. The model is first pretrained on the ZINC 3D database of drug-like molecules to learn general molecular patterns. It is then fine-tuned on CrossDocked2020, a dataset of protein-ligand complexes, to learn pocket-specific generation.
By default, provide a full protein structure in PDB format plus a positioned reference ligand pose in SDF or MOL format. ProteinIQ uses PocketFlow's native pocket-splitting helper to extract residues near the ligand before generation.
The reference ligand is not docked. Its 3D coordinates define the binding site, and native PocketFlow keeps nearby residues using the pocket cutoff setting. The native preprocessing default is 10 Å.
If you already have a pocket PDB, switch Pocket input to Pre-extracted pocket and upload that structure directly. The pocket should contain the protein residues surrounding the binding site where you want molecules generated. We recommend using clean structures without waters or ions unless they're critical for binding.
generated.sdf.10 Å.These advanced settings control the stochastic generation process.
Temperature parameters affect sampling diversity. Lower temperatures produce more conservative, predictable structures while higher temperatures explore more unusual chemical space.
1.0 favor common atom types, above 1.0 increases diversity.Focus parameters control how the model selects which atom to extend next during generation.
PocketFlow returns generated molecules in native generation order. ProteinIQ also calculates QED (Quantitative Estimate of Drug-likeness) and standard molecular properties as derived annotations for easier triage.
PocketFlow does not predict binding affinity or activity. Use downstream docking tools such as AutoDock Vina or GNINA when you need binding-oriented ranking.
QED ranges from 0-1, with higher values indicating more drug-like properties. The score combines molecular weight, lipophilicity, hydrogen bond donors/acceptors, polar surface area, rotatable bonds, and aromatic rings into a single metric.
QED > 0.7: Highly drug-likeQED 0.5-0.7: Moderate drug-likenessQED < 0.5: Less drug-like, may require optimizationDownload individual molecules as SDF files for further analysis in molecular modeling software. The 3D coordinates correspond to the predicted binding pose within the pocket.
Batch library mode returns aggregate files for downstream screening:
The browser viewer reads individual molecule records from generated.sdf when you select a row, so batch library jobs do not create separate molecule_###.sdf preview files.
Use a ligand pose already positioned in the intended binding site. If the ligand pose is misplaced, the extracted pocket will be misplaced too.
Extracted pockets need sufficient context (8-12Å from the binding site center). Too small pockets may constrain generation, while overly large pockets increase noise.
Remove crystallographic artifacts like buffer molecules, and consider whether to keep structural waters based on their role in binding.
Use PocketFlow as part of an iterative design workflow:
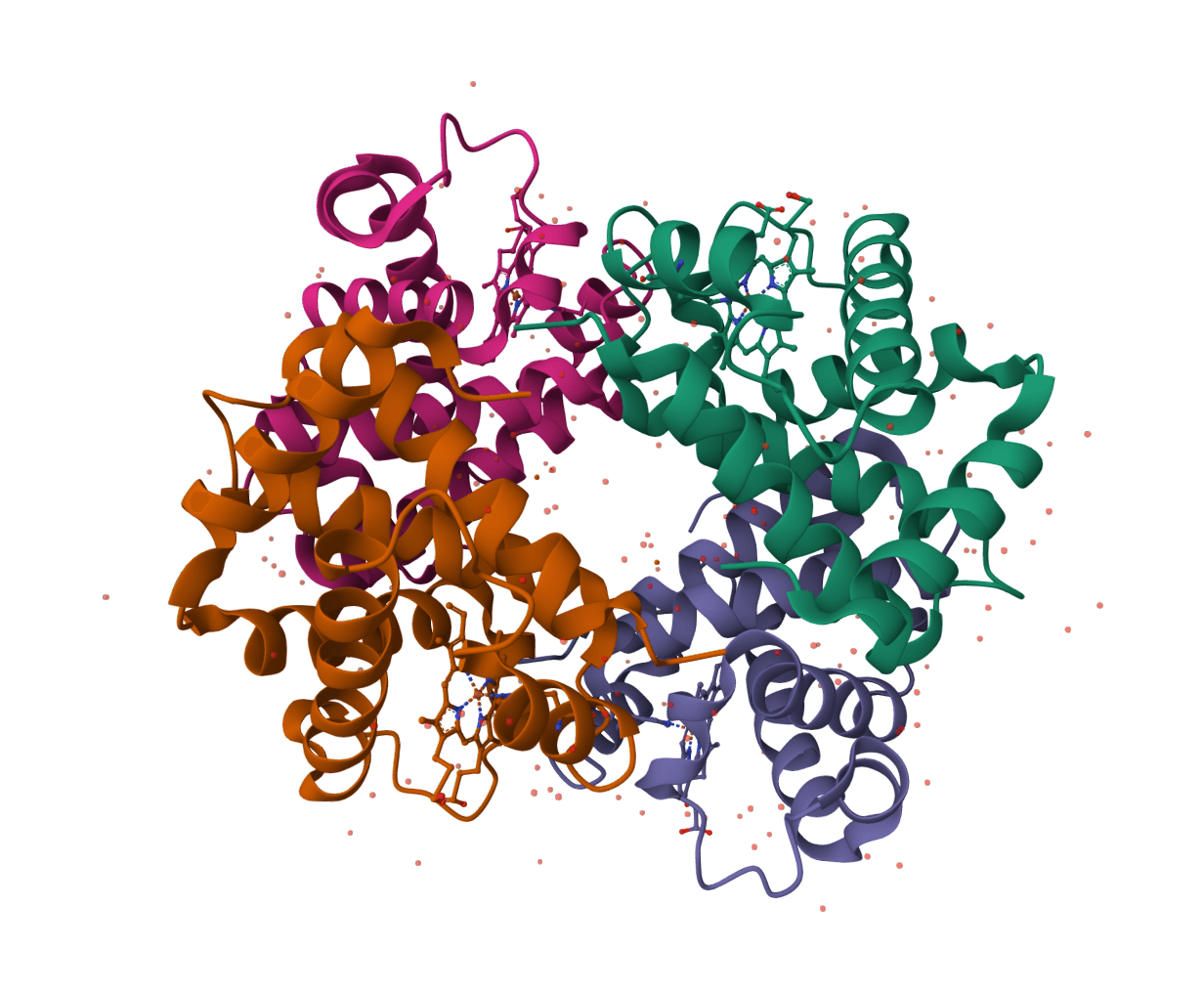
PocketXMol is a pocket-interacting generative foundation model for docking, small-molecule design, and peptide design in protein binding pockets.
GenMol is a generative AI model from NVIDIA that creates novel drug-like molecules using masked discrete diffusion. It generates molecules in SAFE representation format and supports de novo generation, linker design, motif extension, and scaffold decoration.
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
RFdiffusion is a state-of-the-art protein structure generation tool that uses diffusion models to design proteins de novo, create binders, scaffold motifs, and generate symmetric oligomers with atomic precision.
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.
PocketFlow is a structure-based deep generative model that designs novel drug-like molecules inside protein binding pockets. Published in Nature Machine Intelligence in March 2024 by researchers at Sichuan University, it combines autoregressive flow modeling with explicit chemical knowledge to generate molecules with 100% chemical validity.
What sets PocketFlow apart is its experimental validation. The authors applied PocketFlow to design inhibitors for two epigenetic targets (HAT1 and YTHDC1) and successfully obtained wet-lab-validated bioactive lead compounds. This makes PocketFlow the first structure-based molecular deep generative model with experimental validation of designed molecules.
In computational benchmarks on the CrossDocked2020 dataset, PocketFlow outperforms previous methods while maintaining perfect chemical validity and high drug-likeness scores.
PocketFlow generates molecules atom-by-atom within a protein binding pocket using an autoregressive approach. At each step, the model decides what atom type to add, where to place it in 3D space, and how to connect it to existing atoms. Chemical rules guide these decisions to ensure valid molecules.
The core of PocketFlow is the Geometric Double Bottleneck Perceptron (GDBP), an SE(3)-equivariant graph neural network that models the 3D geometry of the protein-ligand complex. GDBP improves upon earlier geometric neural networks (GVP and GBP) by adding bottleneck layers for both scalar and vector features.
This architecture processes 3D coordinates directly while maintaining equivariance to rotations and translations. The model can generate atom positions in 3D space without needing to first predict internal coordinates.
The autoregressive generation uses three specialized components working together:
Atom Flow predicts the type of each new atom (carbon, nitrogen, oxygen, etc.) using a normalizing flow. This probabilistic approach captures the distribution of atom types conditioned on the current molecular state and pocket environment.
Position Predictor determines where to place each new atom in 3D space relative to the binding pocket. The GDBP network encodes spatial relationships between existing atoms, protein residues, and potential placement sites.
Bond Flow predicts connectivity between the new atom and existing atoms using another normalizing flow. This component receives explicit chemical knowledge guidance to ensure reasonable bond patterns.
Unlike purely data-driven approaches, PocketFlow incorporates chemical knowledge directly into the generation process. The bond predictor checks whether proposed bonds satisfy valence rules and reasonable bonding patterns.
If the model proposes an unreasonable bond, it resamples until finding a valid connection. This explicit guidance is critical—ablation studies show that removing chemical constraints significantly degrades both validity and drug-likeness of generated molecules.
PocketFlow uses a two-stage training process. The model is first pretrained on the ZINC 3D database of drug-like molecules to learn general molecular patterns. It is then fine-tuned on CrossDocked2020, a dataset of protein-ligand complexes, to learn pocket-specific generation.
By default, provide a full protein structure in PDB format plus a positioned reference ligand pose in SDF or MOL format. ProteinIQ uses PocketFlow's native pocket-splitting helper to extract residues near the ligand before generation.
The reference ligand is not docked. Its 3D coordinates define the binding site, and native PocketFlow keeps nearby residues using the pocket cutoff setting. The native preprocessing default is 10 Å.
If you already have a pocket PDB, switch Pocket input to Pre-extracted pocket and upload that structure directly. The pocket should contain the protein residues surrounding the binding site where you want molecules generated. We recommend using clean structures without waters or ions unless they're critical for binding.
generated.sdf.10 Å.These advanced settings control the stochastic generation process.
Temperature parameters affect sampling diversity. Lower temperatures produce more conservative, predictable structures while higher temperatures explore more unusual chemical space.
1.0 favor common atom types, above 1.0 increases diversity.Focus parameters control how the model selects which atom to extend next during generation.
PocketFlow returns generated molecules in native generation order. ProteinIQ also calculates QED (Quantitative Estimate of Drug-likeness) and standard molecular properties as derived annotations for easier triage.
PocketFlow does not predict binding affinity or activity. Use downstream docking tools such as AutoDock Vina or GNINA when you need binding-oriented ranking.
QED ranges from 0-1, with higher values indicating more drug-like properties. The score combines molecular weight, lipophilicity, hydrogen bond donors/acceptors, polar surface area, rotatable bonds, and aromatic rings into a single metric.
QED > 0.7: Highly drug-likeQED 0.5-0.7: Moderate drug-likenessQED < 0.5: Less drug-like, may require optimizationDownload individual molecules as SDF files for further analysis in molecular modeling software. The 3D coordinates correspond to the predicted binding pose within the pocket.
Batch library mode returns aggregate files for downstream screening:
The browser viewer reads individual molecule records from generated.sdf when you select a row, so batch library jobs do not create separate molecule_###.sdf preview files.
Use a ligand pose already positioned in the intended binding site. If the ligand pose is misplaced, the extracted pocket will be misplaced too.
Extracted pockets need sufficient context (8-12Å from the binding site center). Too small pockets may constrain generation, while overly large pockets increase noise.
Remove crystallographic artifacts like buffer molecules, and consider whether to keep structural waters based on their role in binding.
Use PocketFlow as part of an iterative design workflow:
PocketXMol is a pocket-interacting generative foundation model for docking, small-molecule design, and peptide design in protein binding pockets.
GenMol is a generative AI model from NVIDIA that creates novel drug-like molecules using masked discrete diffusion. It generates molecules in SAFE representation format and supports de novo generation, linker design, motif extension, and scaffold decoration.
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
RFdiffusion is a state-of-the-art protein structure generation tool that uses diffusion models to design proteins de novo, create binders, scaffold motifs, and generate symmetric oligomers with atomic precision.
RFdiffusion2 is an atom-level enzyme active site scaffolding tool that generates protein scaffolds around your input motif. REQUIRES an input PDB structure containing the active site residues to scaffold. For ligand-aware design, ligands must be embedded in the input PDB as HETATM records.