Alignment
ANARCI
Number antibody and T cell receptor variable domain sequences using multiple numbering schemes (IMGT, Chothia, Kabat, Martin, AHo, Wolfguy). Identifies chain type, species, and assigns germline genes.
Clustal Omega
Perform multiple sequence alignment on protein or nucleotide sequences using the Clustal Omega algorithm.
FastTree
Infer approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences.
FoldSeek
Fast protein structure search, comparison, and clustering. Search your structure against 200M+ AlphaFold predictions, compare 2 structures, or cluster up to 2500.
HMMER
Sensitive sequence homology search using profile hidden Markov models. More accurate than BLAST for detecting remote homologs, ideal for finding evolutionarily distant protein family members.
IgBLAST
Analyze immunoglobulin (antibody) and T cell receptor variable domain sequences. Identifies V/D/J gene segments, delineates CDR regions, and analyzes rearrangement junctions.
IQ-TREE
Build phylogenetic trees using maximum likelihood with automatic model selection (ModelFinder) and ultrafast bootstrap support.
MAFFT
Perform multiple sequence alignment using MAFFT (Multiple Alignment using Fast Fourier Transform). Supports multiple algorithms from fast progressive to highly accurate iterative methods.
MMseqs2
Ultra-fast sequence search and clustering. 10,000x faster than BLAST for database searches, with powerful sequence clustering capabilities for proteins and nucleotides.
MUMmer4
Rapidly align and compare DNA sequences using MUMmer4 nucmer. Perform pairwise genome comparisons to identify SNPs, indels, and structural variants between reference and query genomes.
MUSCLE5
Perform multiple sequence alignment using MUSCLE5 (MUltiple Sequence Comparison by Log-Expectation). Uses the PPP algorithm for high-quality alignments with support for ensemble generation.
RNAalifold
RNAalifold computes consensus RNA secondary structure from a multiple sequence alignment. Uses covariation information to improve prediction accuracy for evolutionarily conserved structures.
Salmon
Quantify transcript abundance from RNA-seq reads with Salmon selective alignment. Upload a transcript FASTA reference plus single-end or paired-end FASTA/FASTQ reads to produce TPM and estimated read-count tables.
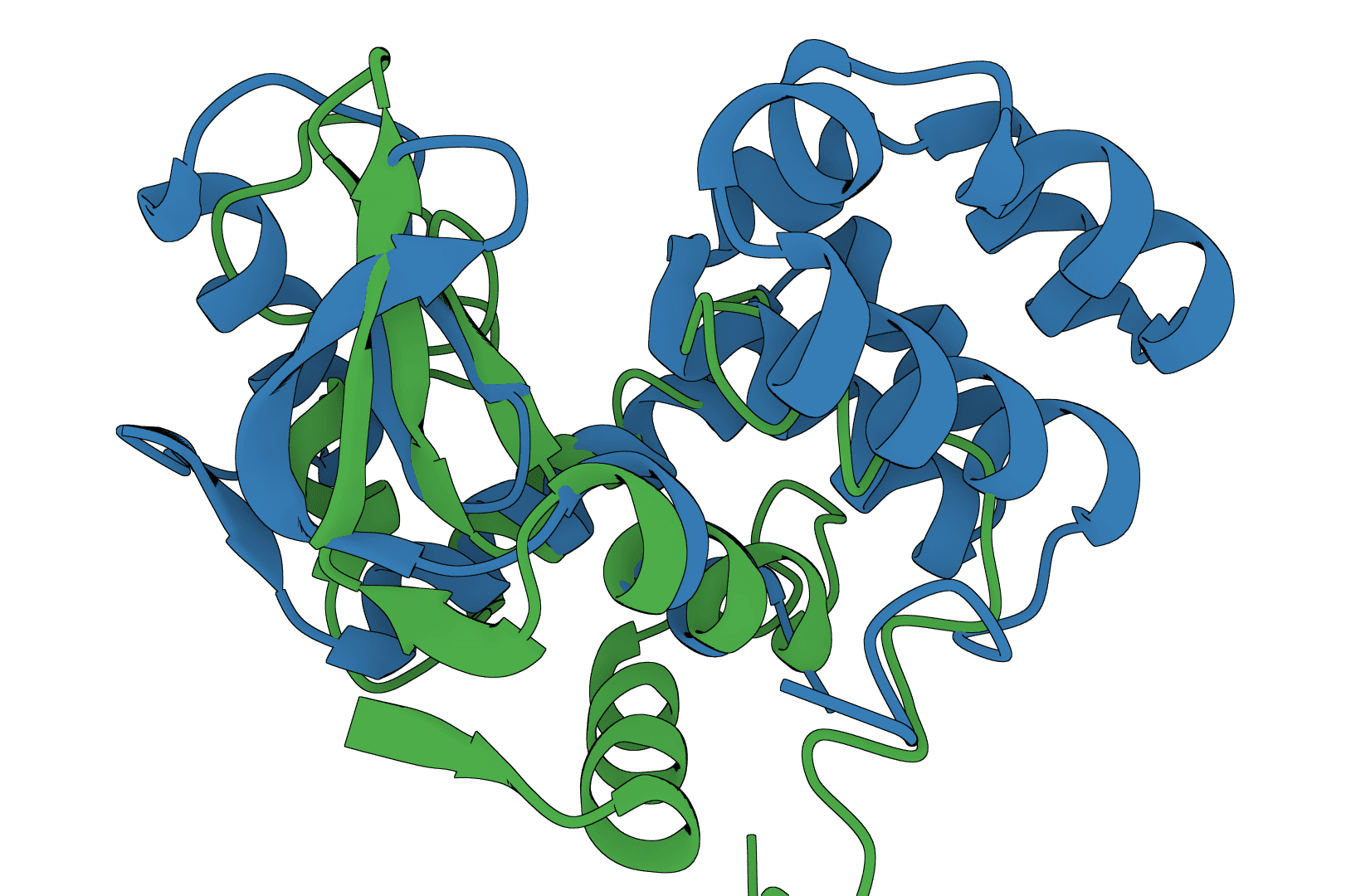
USAlign
USAlign (Universal Structure Alignment) aligns protein, RNA, and DNA structures to compute TM-scores and generate superposed structures. Compare 3D structures to assess structural similarity.