PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
Design linear peptide binders for target proteins using a target sequence-conditioned masked language model. PepMLM generates peptide sequences optimized to bind specific protein targets based on ESM-2 protein language modeling.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
Structure-based de novo antibody and nanobody design pipeline combining antibody-tuned RFdiffusion, ProteinMPNN sequence design, and antibody-tuned RoseTTAFold2 filtering.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
Design protein binders against a target structure with NVIDIA BioNeMo's Proteina-Complexa generative pipeline.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
PepMimic designs short peptides that mimic the binding interface of a known protein binder on its target. From a reference protein complex, a latent diffusion model generates peptide candidates constrained to the target interface, and each candidate is scored by interface-mimicry against the reference binder.
Design linear peptide binders for target proteins using a target sequence-conditioned masked language model. PepMLM generates peptide sequences optimized to bind specific protein targets based on ESM-2 protein language modeling.
PocketXMol is a pocket-interacting generative foundation model for small-molecule or peptide docking and design in protein binding pockets.
Structure-based de novo antibody and nanobody design pipeline combining antibody-tuned RFdiffusion, ProteinMPNN sequence design, and antibody-tuned RoseTTAFold2 filtering.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
Design protein binders against a target structure with NVIDIA BioNeMo's Proteina-Complexa generative pipeline.
EvoDiff is a diffusion-based protein sequence generation framework from Microsoft Research. ProteinIQ currently runs the EvoDiff-Seq OA_DM_38M model for unconditional protein generation, motif scaffolding, and user-sequence inpainting.
Generate protein structures and scaffolds with Genie 3, an all-atom SE(3)-equivariant diffusion model. Genie 3 supports unconditional protein generation, motif scaffolding, and hotspot-targeted binder design.
All-atom generative AI for designing protein binders. Specify target binding sites and generate diverse binding proteins with fine-grained control over interaction parameters.
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Peptide binder (12 residues)
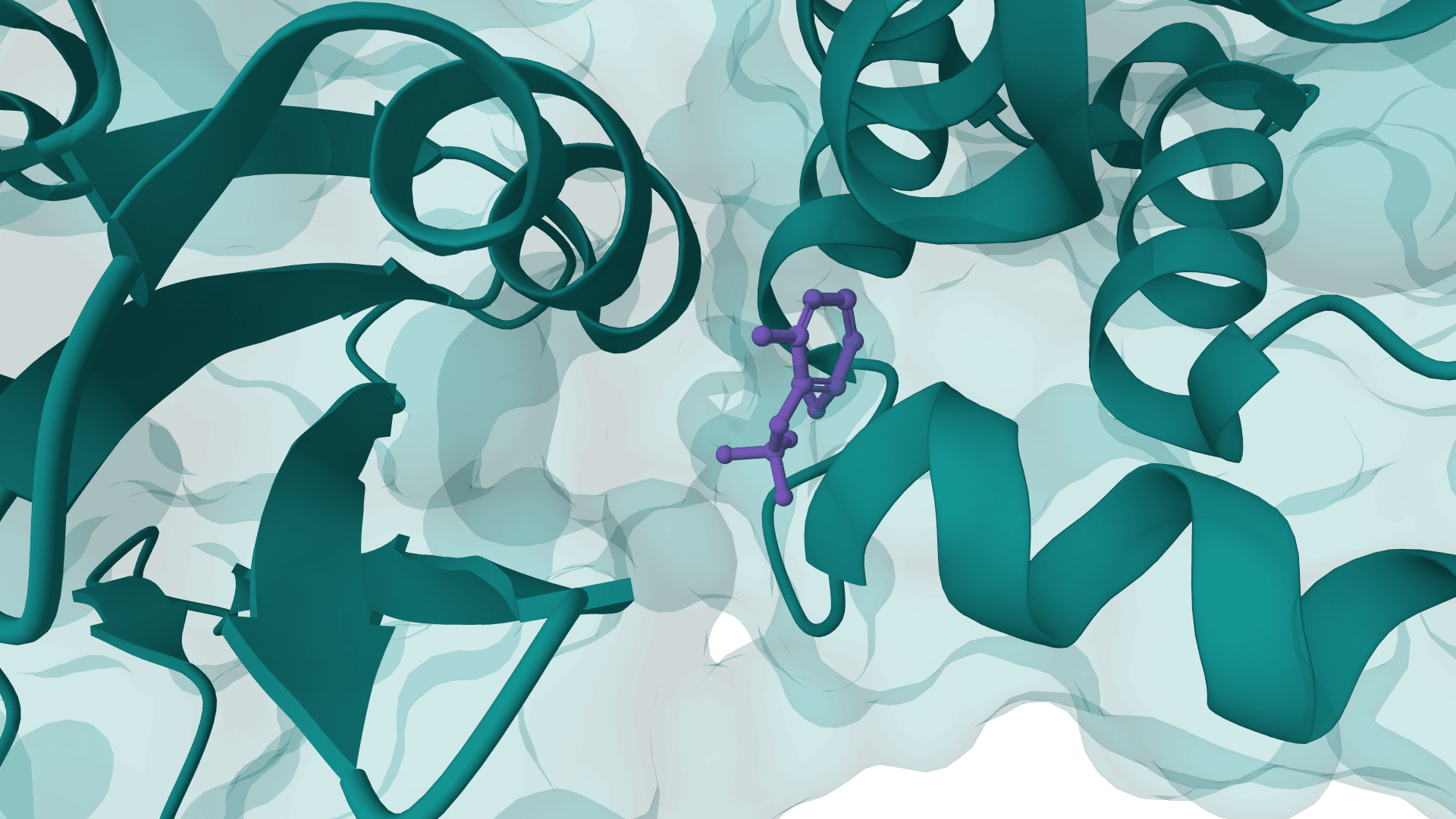
BoltzGen is an all-atom diffusion model for binder design. It supports de novo peptide and protein binders, structure redesign, nanobody and Fab scaffold design, and protein design around a small-molecule target.
ProteinIQ runs the native BoltzGen v0.3.2 pipeline on hosted GPU infrastructure. Each job generates a fresh, complete campaign and returns the native structures, metrics, plots, and run configuration needed to review the result.
| Input | Description |
|---|---|
Target protein | A .pdb, .ent, .cif, or .mmcif structure, or an RCSB PDB ID. CIF/mmCIF content is passed to BoltzGen without conversion so canonical chain and residue identifiers are preserved. |
Nanobody framework | A custom VHH framework for custom-framework nanobody design. Required only when Upload my framework is selected. |
Target ligand | A SMILES string, CCD code, or PubChem result for Protein-small molecule mode. |
For mmCIF inputs, chain and residue settings use label_asym_id and 1-based label_seq_id values. These can differ from author-provided identifiers.
| Protocol | Designed entity |
|---|---|
Peptide | A short linear or cyclic peptide against a structure target. |
Protein | A de novo protein binder against a structure target. |
Protein redesign | Selected regions of an uploaded protein complex. |
Nanobody | CDR regions in a bundled or uploaded VHH framework. |
Antibody / Fab CDR | CDR regions in a selected therapeutic Fab scaffold. |
Protein-small molecule | A de novo protein around a SMILES or CCD ligand. |
Binder length controls appear only for direct peptide, protein, and protein-small-molecule design. Nanobody and Fab lengths come from their native scaffold files; redesign length comes from the uploaded structure.
| Setting | Description |
|---|---|
Number of designs | Candidate count before filtering. Hosted runs support 1–100 candidates per job. Runtime scales with this value. A single design is useful for a quick trial. |
Budget | Maximum number of final designs retained by native quality-and-diversity selection. It cannot exceed the generated candidate count. |
Uniform binder size | Uses one exact direct-binder length. Disabling it samples an inclusive minimum-to-maximum range. |
Long campaigns can end before every requested candidate finishes. Completed BoltzGen designs and native artifacts produced before execution stops are returned when available.
The BoltzGen source documentation discusses campaigns containing thousands of candidates. ProteinIQ supports quick single-design trials and campaigns of up to 100 candidates per job. Broader campaigns should use several independent jobs and compare their native metric tables.
| Setting | Description |
|---|---|
Binding site | Target residues in chain:residue_spec form, such as A:12,14,61. |
Target chains | Comma-separated target chains to include, such as A,B. |
Redesign regions | Regions in native chain:residue_spec form, such as A:10..20 or B:42,45,50..55. |
Framework chain | The VHH chain in an uploaded custom nanobody framework. |
CDR design regions | Closed, 1-based canonical ranges such as 26..34,52..59,98..118. |
Sample CDR lengths | Enables native deletion and insertion sampling for selected segments inside the custom CDR design regions. |
Variable CDR segments | Removed residues and replacement lengths in removed_start..removed_end:min..max form, such as 26..28:1..5. |
Custom-framework mode preserves the uploaded framework and redesigns only the selected CDR regions. CDR lengths remain fixed by default. When length sampling is enabled, each configured segment is removed and replaced with a length sampled from its supplied range. Every removed segment must lie entirely inside one CDR design region. ProteinIQ does not infer antibody numbering or CDR boundaries.
These controls apply to direct designed protein entities. Malformed or mode-inapplicable constraints fail before compute starts instead of being silently ignored.
| Setting | Syntax |
|---|---|
Secondary structure | chain:start-end:HELIX, SHEET, or LOOP |
Disulfide bonds | chain:residue,chain:residue |
Staple bonds | chain:residue:atom,chain:residue:atom |
Fixed sequence regions | chain:start-end:sequence |
Binding residues | chain:residue1,residue2 |
Non-binding residues | chain:residue1,residue2 |
Residue constraints | chain:position:allowed|disallowed:amino_acids |
Design insertions | chain:position:min..max[:secondary_structure], for protein redesign only |
Fixed sequence regions use BoltzGen's native mixed fixed/designable sequence notation. They require a uniform binder length and cannot overlap. The direct binder chain is B for peptide/protein target modes and A for protein-small-molecule mode.
| Setting | Description |
|---|---|
Skip inverse folding | Skips sequence redesign and evaluates the generated design representation directly. |
Sequences per backbone | Number of inverse-folded sequence variants per generated backbone. |
Avoid amino acids | Standard one-letter amino-acid codes excluded during inverse folding. An empty value preserves BoltzGen's protocol policy. |
Quality vs diversity (alpha) | Optional final-selection override. Empty preserves the native default: 0.01 for peptide design and 0.001 for other protocols. |
Refolding RMSD threshold | Optional filtering override. Empty preserves the native threshold: 2.0 Å for peptides and 2.5 Å for other protocols. |
Step scale | Optional diffusion override. Empty preserves the native per-stage schedule. |
Noise scale | Optional diffusion override. Empty preserves the native per-stage schedule. |
Custom filters | One strict metric<value or metric>value expression per line. |
Metrics weights | One metric=value entry per line. Use metric=none to remove a metric from ranking. |
Size buckets | One min-max:count cap per line. |
Each hosted job uses a new run directory, so resume, partial-stage, and inverse-fold-only controls are intentionally not offered. Those source controls require compatible persistent intermediates or a prepared binder-target structure.
| Output | Description |
|---|---|
Ranked complex CIF | Native refolded binder-target complex for each retained design. |
Binder-only CIF | Native binder-only refold for each retained design when produced by the protocol. |
Native metric tables | Final, all-design, aggregate, and per-target CSV files when produced. |
Results overview | Native BoltzGen PDF plots when produced. |
Run provenance | Native steps.yaml and generated configuration YAML files. |
Reference inputs | Uploaded target and custom framework files, or the ligand reference. |
BoltzGen's standard pipeline does not produce FASTA files. Designed sequences are available in the ranked table and native CSV files.
| Column | Native meaning |
|---|---|
Binder pTM | design_ptm, the predicted quality of the designed component. It is not whole-complex pTM. |
Binder–target iPTM | design_to_target_iptm, interface confidence between the design and target. |
Minimum binder–target PAE (Å) | min_design_to_target_pae; lower values indicate more confident relative placement. |
Delta SASA (Ų) | delta_sasa_refolded when available; larger values indicate more buried surface after complex formation. |
Normalized rank score | Native quality_score used in final ranking. |
Sequence | Native designed amino-acid sequence for the retained candidate. |
Always inspect the raw metric tables. Metric usefulness and acceptable thresholds depend on protocol, target flexibility, binder class, and campaign design; no single displayed score establishes experimental binding.
A complete hosted run follows BoltzGen's native stages:
Protein-small-molecule runs also include affinity-oriented analysis. Protein redesign skips de novo binder generation but still performs complex folding, analysis, and filtering.