Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.

Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Download AlphaFold DB predicted structures and confidence files by UniProt accession.
Fix ligand files that fail RDKit, Meeko, or docking preparation. Repair SDF, MOL, and MOL2 inputs, apply safe chemistry cleanup, and export docking-ready SDF files.
Download PDB, CIF, and FASTA files from RCSB PDB by 4-character structure ID.
PDB2PQR prepares protein structures for electrostatics calculations by adding missing atoms, predicting protonation states using PROPKA, and assigning atomic charges and radii from standard force fields.
Download PDBe structure files as PDB and CIF by PDB ID.
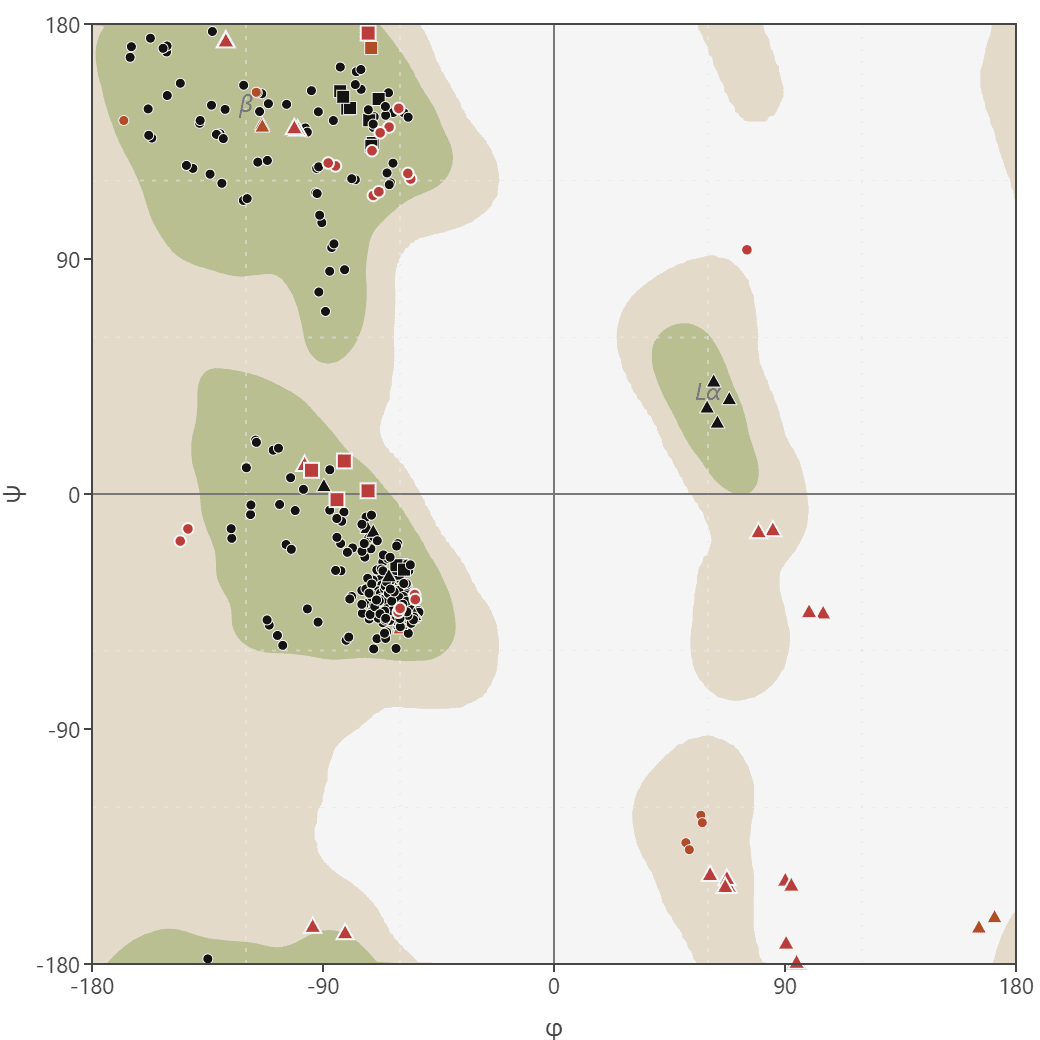
Generate Ramachandran plots from PDB or mmCIF structures to analyze protein backbone dihedral angles (phi/psi). Visualize favored, allowed, and outlier regions.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Clean and filter DNA sequences by removing or replacing non-standard nucleotide characters. Supports multiple filter modes including standard 4 bases, IUPAC ambiguity codes, and custom character sets.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Download AlphaFold DB predicted structures and confidence files by UniProt accession.
Fix ligand files that fail RDKit, Meeko, or docking preparation. Repair SDF, MOL, and MOL2 inputs, apply safe chemistry cleanup, and export docking-ready SDF files.
Download PDB, CIF, and FASTA files from RCSB PDB by 4-character structure ID.
PDB2PQR prepares protein structures for electrostatics calculations by adding missing atoms, predicting protonation states using PROPKA, and assigning atomic charges and radii from standard force fields.
Download PDBe structure files as PDB and CIF by PDB ID.
Generate Ramachandran plots from PDB or mmCIF structures to analyze protein backbone dihedral angles (phi/psi). Visualize favored, allowed, and outlier regions.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Clean and filter DNA sequences by removing or replacing non-standard nucleotide characters. Supports multiple filter modes including standard 4 bases, IUPAC ambiguity codes, and custom character sets.
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
HIV-1 protease default repair
HIV-1 protease docking cleanup
Ubiquitin pH 6.5 repair
PDBFixer repairs PDB and mmCIF structure files in preparation for molecular dynamics simulations. Part of the OpenMM toolkit, it adds missing heavy atoms, hydrogens, and entire loops; converts modified residues to standard amino acids; selects single conformations from alternate locations; deletes unwanted chains; and builds explicit solvent boxes or lipid membranes around the system.
Experimental structures from X-ray crystallography and cryo-EM rarely arrive simulation-ready. They lack hydrogens, contain incomplete side chains from regions of poor electron density, include crystallization artifacts like buffers and duplicate chains, and frequently carry modified residues (selenomethionine, phosphoserine) that standard force fields cannot parameterize. GROMACS, AMBER, and OpenMM all reject the raw files. PDBFixer addresses these issues through eight correction passes:
ProteinIQ accepts up to 10 structures per job and returns one fixed PDB per input with a combined run summary and applied-settings JSON file.
Upload a PDB or mmCIF file (or paste an RCSB PDB ID) to receive a repaired structure with missing atoms, hydrogens, and loops filled in. PDBFixer runs on cloud hardware with no local Python or OpenMM installation. Up to 10 structures can be submitted in a single job, each producing its own fixed PDB file and a shared run summary.
Upload structure files or enter RCSB PDB IDs (1HSG for HIV-1 protease is a common test case). PDB (.pdb, .ent) and mmCIF/PDBx (.cif, .mmcif, .pdbx) formats are accepted, up to 50 MB per file. A single job holds up to 10 structures, all of which share the same repair settings.
Under "Core processing," keep Preparation preset set to Source defaults for source-faithful behavior, or choose MD preparation to standardize modified residues and use 0.15 M salt for solvent or membrane setup. Keep Heterogens set to Keep all to preserve ligands, ions, and water. To extract a single chain from a multi-chain complex, list the unwanted chain IDs in Remove chains (e.g., B, C).
Under "Add options," Add missing heavy atoms and Add missing hydrogens are enabled by default. Both are required for MD simulations. Enable Add missing residues only when complete chains are needed; this step is slow for structures with large gaps.
For explicit solvent MD, enable Add solvent box; the default adds counterions needed for neutralization without extra salt. Set ionic strength to 0.15 M for physiological salt. For membrane proteins, enable Add lipid membrane instead; solvent and membrane are mutually exclusive.
Click Submit job to start processing. Once complete, each repaired PDB can be downloaded directly or opened in the integrated PDB Viewer. A summary CSV covers the entire run, and a provenance JSON file records the requested settings and the resolved settings applied to each structure.
Each input structure is processed independently and the output list preserves the original input order. When names are available, ProteinIQ keeps the original file names or RCSB identifiers for each repaired structure. If the incoming label is only a placeholder such as PDB 1, the platform attempts to recover a more useful name from the uploaded file name, the fetched identifier, or the PDB/mmCIF content itself.
| Input | Description |
|---|---|
PDB | A PDB or mmCIF file (.pdb, .ent, .cif, .mmcif, .pdbx), up to 50 MB. Up to 10 structures per job. PDB IDs fetch directly from RCSB. |
pH value: Sets protonation states for titratable groups when adding hydrogens. Default 7.0. The field only applies when "Add missing hydrogens" is enabled.Preparation preset: Source defaults preserves PDBFixer defaults. MD preparation enables nonstandard residue replacement and sets ionic strength to 0.15 M for simulation setup.Heterogens: Keep all preserves ligands, ions, and water from the original structure. Keep only water removes ligands and ions but preserves crystallographic waters. Remove all strips everything except the protein chains.Remove chains: Comma-separated chain IDs to exclude (e.g., B, C, D).Apply mutations: Point mutations introduced during processing. Format CHAIN:ORIGINAL-POSITION-NEW (e.g., A:ALA-57-GLY, B:VAL-123-ILE).These settings control which structural elements PDBFixer adds to the input.
Add missing residues: Builds entire missing loops from SEQRES records. Off by default. Slow for structures with large gaps (10+ residue loops).Add missing heavy atoms: Completes truncated side chains. On by default. Required for most MD force fields.Add missing hydrogens: Adds hydrogens at the specified pH. On by default. Required for explicit hydrogen MD; can be disabled for rigid-body docking or implicit hydrogen force fields.Replace nonstandard residues: Converts modified amino acids (selenomethionine, phosphoserine) to their standard equivalents. Off by default to preserve deposited chemistry; enable it when standard force-field preparation requires standard residues.Adding explicit water creates a simulation-ready system. The protein is surrounded by a rectangular or truncated box of water molecules with counterions for charge neutralization.
Add solvent box: Enables water box construction. Significantly increases processing time and output file size.Cation/Anion: Ion types for charge neutralization. Na+/Cl- is standard for most simulations. K+ is appropriate where potassium is physiologically relevant.Ionic strength: Molar concentration of added salt beyond the counterions needed to neutralize the system. Default 0.0 M matches PDBFixer; 0.15 M is common for physiological salt.Box geometry: Shape of the periodic boundary box when automatic padding is used. Cubic is standard and compatible with all software. Dodecahedron and Octahedron reduce water count by approximately 30% while maintaining minimum distance from protein to box edge, producing faster simulations with equivalent accuracy.Box sizing: Automatic padding (distance from protein to box edges) or explicit X/Y/Z dimensions in nanometers.Water model: TIP3P is the standard choice for most force fields. TIP4P-Ew provides improved density and diffusion properties. SPC/E is popular for GROMACS workflows.Membrane systems embed the protein in a lipid bilayer for studying GPCRs, ion channels, and transporters.
Add lipid membrane: Constructs a membrane system. Cannot be combined with Add solvent box; the membrane system includes water and ions automatically.Lipid type: Composition of the bilayer. POPC (palmitoyl-oleoyl-phosphatidylcholine) is the standard choice for general membrane protein studies. POPE is preferred for bacterial membranes.Membrane center Z: Position of the bilayer center along the Z-axis in nanometers. Set to 0.0 for automatic centering. Adjust for asymmetric placement in the membrane.Minimum padding: Distance from the protein to the box edges in nanometers.Force field: Molecular mechanics force field for atom placement and minimization. AMBER14 is the standard default. CHARMM36 is preferred for simulations using CHARMM-compatible software.Random seed: Specific integer for reproducible structure generation. 0 produces random output. Fixed seeds guarantee identical output when reprocessing the same structure with the same settings.Use custom box vectors: Manual triclinic box vectors (A, B, C as comma-separated X,Y,Z components in nanometers) instead of standard box shapes. Useful for non-orthogonal simulation setups.Download templates: Comma-separated residue codes to fetch from the PDB Chemical Component Dictionary (e.g., ATP, GTP, HEM). Use for non-standard residues not included in the default template library.PDBFixer applies a stepwise workflow that compares each residue against template records and uses OpenMM's force fields to place missing atoms in chemically reasonable positions. It packages the most common preparation steps behind a single streamlined path.
For each residue, PDBFixer consults the PDB Chemical Component Dictionary to determine which atoms should exist. Missing atoms are placed at ideal bond lengths, angles, and dihedrals defined in the template library. The deposited coordinates of resolved atoms are preserved exactly; only missing pieces are reconstructed.
After atoms are placed, brief energy minimization using OpenMM's force fields resolves steric clashes between newly added atoms and the existing structure. This relaxation is local, so experimental coordinates shift only slightly. Full equilibration is still required downstream.
When "Add missing hydrogens" is enabled, the pH value setting determines protonation. At pH 7.0, histidine residues are added neutral, aspartate and glutamate deprotonated, lysine and arginine protonated. Disulfide bonds are detected automatically and the bonded cysteines are kept neutral. For atypical pH (lysosomal pH 4.5, endosomal pH 5.5), adjust the setting accordingly; PROPKA can be run first to identify residues with shifted pKa values that warrant special attention.
Missing residues are reconstructed using fragment-based loop building followed by minimization. The algorithm reads SEQRES records to identify the expected residue sequence, places the missing stretch with idealized geometry, and refines with energy minimization. Long gaps (10+ residues) often need additional equilibration to settle into reasonable conformations.
Each input produces one fixed PDB file. A combined summary CSV records the source name, success status, output filename, atom and residue counts, processing steps applied, and any error message for structures that could not be repaired. The pdbfixer_settings_provenance.json file records tool versions, requested settings, and the resolved settings applied to each structure.
| Metric | Description |
|---|---|
Source | Original file name or RCSB identifier |
Atoms | Total atom count in the fixed structure |
Residues | Residue count (increases when loops are added) |
Chains | Number of protein chains retained |
Processing applied | List of fixes performed, e.g., "Added 2,847 hydrogens, replaced 3 nonstandard residues" |
The 3D viewer labels each repaired model by its source name and shows one structure at a time, since PDBFixer does not generate alternative poses. Ligand-specific grouping controls are hidden for the same reason.
Visual inspection before long simulations catches problems that propagate into MD trajectories:
If added loops look unrealistic, extended energy minimization or brief MD equilibration refines the structure. The PDB Viewer provides in-browser inspection, and MolProbity delivers detailed geometry validation.
Routine structure preparation in PyMOL, Chimera, or Swiss-PdbViewer involves a sequence of manual steps: identify missing atoms, fetch templates, set protonation states, build solvent, neutralize charges. PDBFixer packages these steps into a single deterministic pipeline.
| Aspect | PDB Fixer | Manual preparation |
|---|---|---|
| Input formats | PDB and mmCIF/PDBx | Varies by software |
| Missing atoms | Automatic detection and placement | Requires scripting or plugins |
| Hydrogens | pH-dependent protonation states | Often uniform or default states |
| Loop building | Automated with minimization | Requires homology modeling tools |
| Large structures | Cloud CPU processing with tiered atom limits | CPU-only in most tools |
| Reproducibility | Deterministic with fixed seed | Depends on operator |
| Processing time | Minutes for typical structures; large repairs can take longer | Hours for complex cases |
| Learning curve | Minimal | Requires software expertise |
PDBFixer is well suited to routine multi-structure processing and simulation setup. For complex cases requiring manual intervention (unusual ligands, specific protonation states, asymmetric membrane positioning), it serves as a strong starting point that can be refined in dedicated tools.
Several tools overlap with PDBFixer on specific preparation tasks.
Modeller is a lower-level API used by this workflow. It provides more granular control (custom residue templates, hybrid solvent setups) but requires Python scripting. Use it when the standard PDBFixer settings do not capture a required preparation step.tleap is the standard preparation tool for AMBER force fields. It integrates directly with AMBER-specific parameter sets; the trade-off is less automation than PDBFixer.For protonation state prediction at a specific pH, PROPKA provides pKa values that can guide the pH setting passed to PDBFixer. For binding site analysis on the fixed structure, fpocket identifies druggable pockets; for protein-ligand docking on the cleaned structure, AutoDock Vina and DiffDock are the standard starting points.
Select the MD preparation preset, then add a solvent box with 1.0 nm padding when explicit solvent is needed. The preset keeps missing heavy atoms and hydrogens enabled, standardizes modified residues, and uses 0.15 M salt. The output PDB includes box vectors and counterions, ready for equilibration in OpenMM, GROMACS, or AMBER.
Fix the structure with default settings, then use AutoDock Vina or DiffDock for molecular docking. For docking, switching Heterogens to Remove all strips water but preserves crystallographic ligands as reference poses.
Structures from ESMFold, Boltz-2, AlphaFold 2, or OpenFold 3 often need protonation or format adjustments before downstream analysis. PDBFixer adds hydrogens and standardizes the structure.
Submit up to 10 structures in a single job, useful for cleaning a homology model series or preparing a virtual screening library. Each entry produces its own fixed PDB; a shared summary CSV tracks the entire run.
Add missing heavy atoms and Add missing hydrogens handle most preparation needs without aggressive modification.Add missing residues unless complete chains are required. Long gap fills produce approximate conformations needing extensive equilibration. If the missing region is not relevant to the study, leave it missing.0 M for neutralization-only preparation, use 0.15 M for physiological salt, and consider 0.5 to 1.0 M for halophiles or aggregation studies.TIP3P works with AMBER14 and CHARMM36; SPC/E is common in GROMACS workflows. Mixing incompatible water models with force fields produces incorrect thermodynamic properties.POPC for general eukaryotic membranes, POPE for bacterial inner membranes.PDBFixer is open-source software licensed under MIT/LGPL. ProteinIQ provides a web interface with a free account tier and paid tiers for larger structures.
Small proteins (under 5,000 atoms) with default settings often finish quickly. Larger structures, missing-residue rebuilding, and solvent or membrane construction take longer on CPU, and structures with extensive loop building or large solvent boxes can take several minutes or more.
PDBFixer builds loops with idealized geometry followed by brief energy minimization. Long loops (10+ residues) often need additional refinement, such as extended minimization or short MD equilibration, to relax into reasonable conformations.
PDBFixer adds missing atoms and prepares structures for simulation. PROPKA predicts pKa values for titratable residues without modifying the structure. The two are complementary: running PROPKA first identifies residues with shifted pKa values, and the pH setting passed to PDBFixer is then adjusted accordingly.
Yes, with Add solvent box enabled. The output includes periodic box vectors and is ready for equilibration in OpenMM, GROMACS, or AMBER.
The Heterogens setting controls this. Remove all strips all non-protein molecules, including ligands. Keep all preserves them along with crystallographic waters.
Replace nonstandard residues converts modified residues to their standard equivalents when enabled. It is off by default to preserve modifications, but standard force fields may not parameterize them. Download templates can fetch specific residue definitions from the PDB Chemical Component Dictionary for custom ligands.
AMBER14 is the standard default and works for most simulations. CHARMM36 is preferred for downstream CHARMM-compatible software and certain lipid or nucleic acid systems.
Yes. PDBFixer processes all chains in the input structure. Remove chains excludes specific chains from the output.
Enable Add lipid membrane rather than Add solvent box. Select the appropriate lipid type (POPC for general eukaryotic membranes, POPE for bacterial inner membranes) and set the membrane center to position the transmembrane domain correctly.