Run
5 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
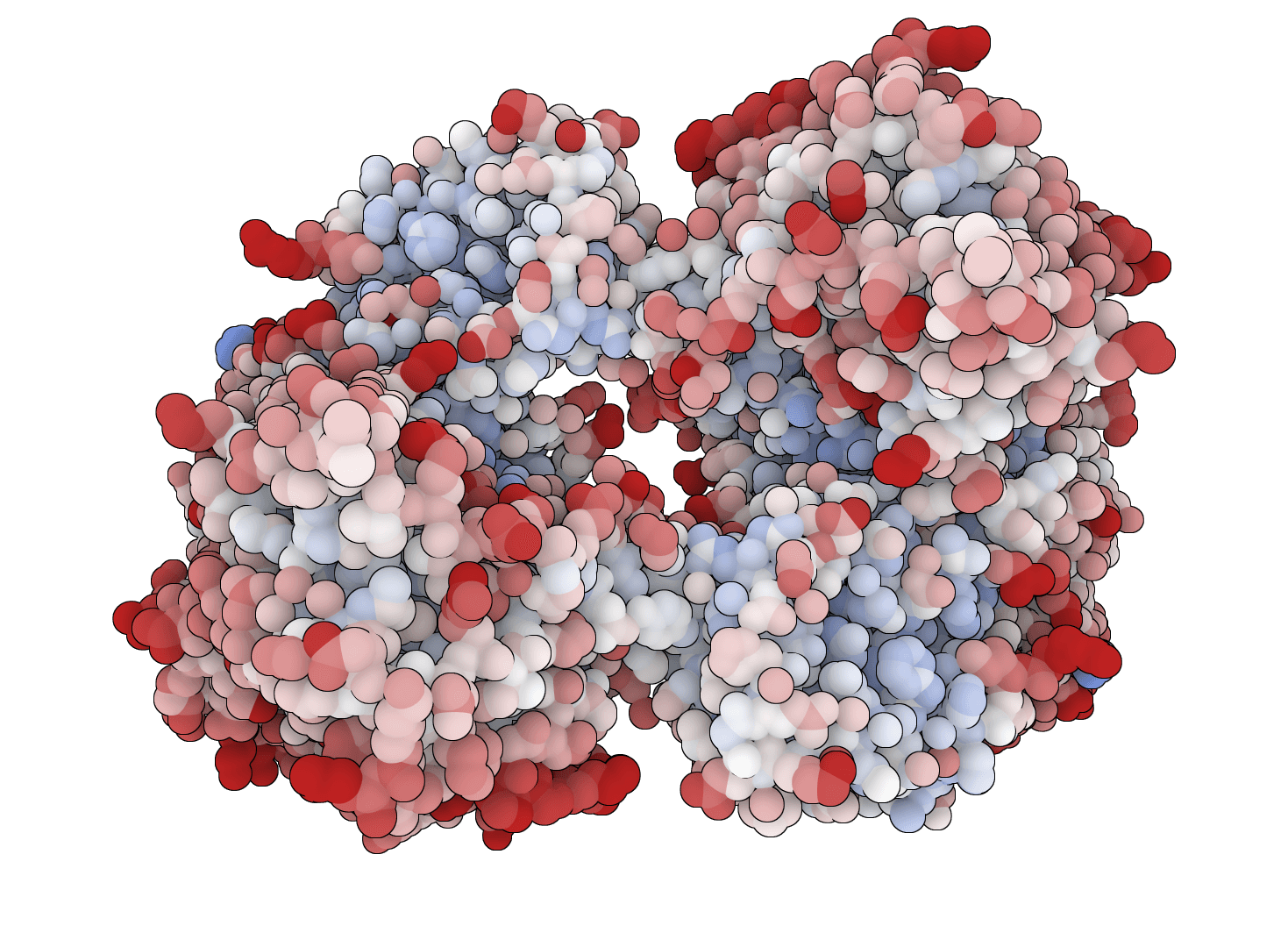
Ubiquitin · complete pKa profile
Lysozyme catalytic dyad · selected residues
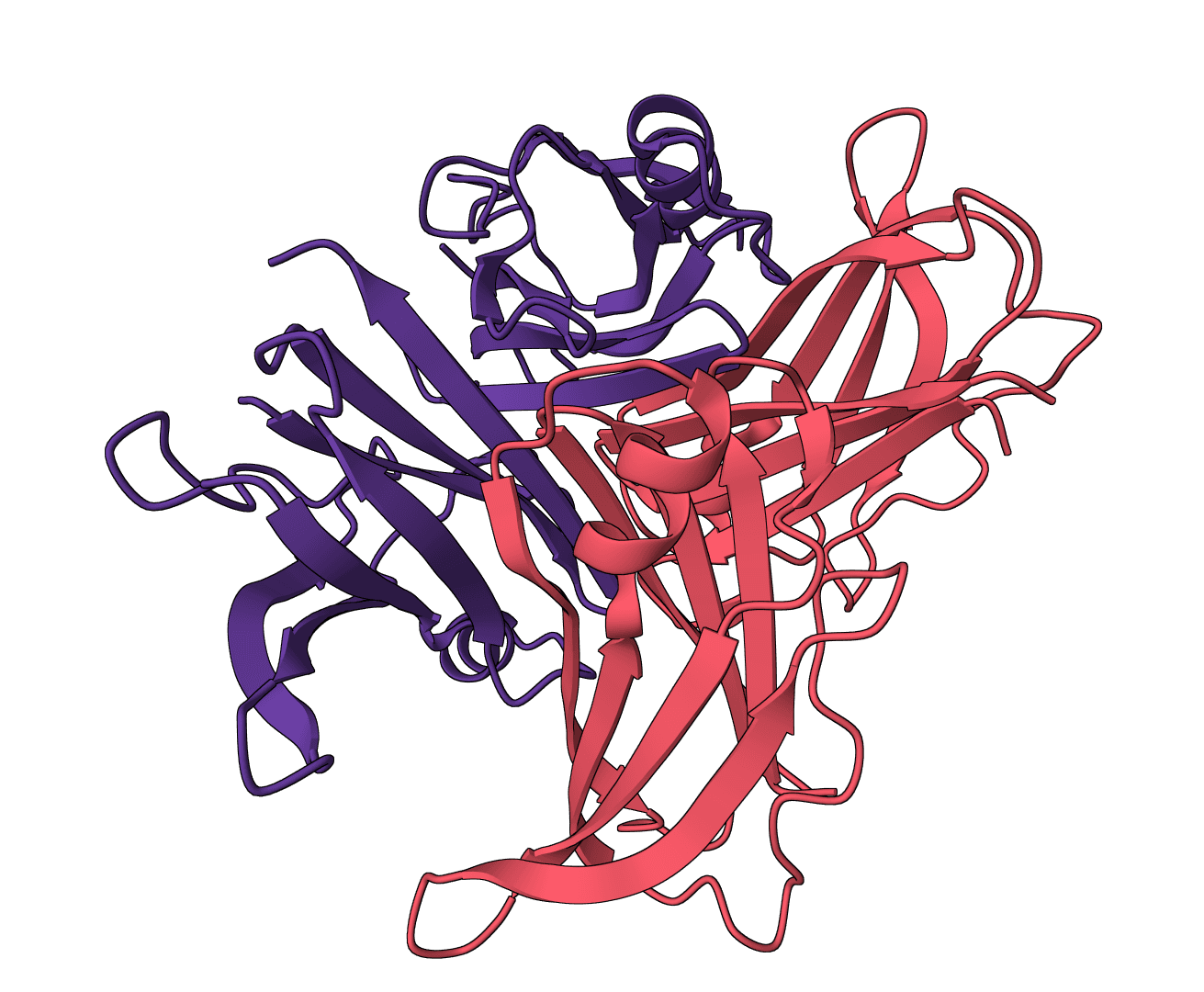
HIV-1 protease–MK1 complex · ligand sites
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Ubiquitin · complete pKa profile
Lysozyme catalytic dyad · selected residues
HIV-1 protease–MK1 complex · ligand sites
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Ubiquitin · complete pKa profile
Lysozyme catalytic dyad · selected residues
HIV-1 protease–MK1 complex · ligand sites
PROPKA is an empirical method for predicting the pKa values of ionizable amino acid residues in proteins based on their three-dimensional structure. Developed by Jan H. Jensen and colleagues at the University of Copenhagen, PROPKA calculates how the protein environment shifts the pKa of each ionizable group away from its intrinsic (model) value.
The pKa of an ionizable group determines at what pH that group gains or loses a proton. In solution, amino acids have characteristic pKa values, but within a folded protein, factors such as burial from solvent, hydrogen bonding, and electrostatic interactions with nearby charged residues can shift these values substantially. PROPKA quantifies these perturbations using structure-based empirical energy functions, enabling rapid prediction of protonation states at any given pH.
PROPKA 3 introduced consistent treatment of internal and surface residues through improved desolvation and dielectric response modeling. The method handles ASP, GLU, HIS, CYS, TYR, LYS, ARG residues and both N- and C-terminal groups. Benchmarks show root-mean-square deviations of 0.79 pKa units for Asp/Glu, 0.75 for Tyr, 0.65 for Lys, and 1.00 for His residues compared to experimental measurements.
ProteinIQ provides a web-based interface for running PROPKA 3.5.1 without command-line installation. Upload a protein structure file or fetch one from RCSB, and receive predicted pKa values for ionizable residues and supported ligand groups present in the structure.
| Input | Description |
|---|---|
Protein Structures | The target protein structure. Upload a PDB file (.pdb or .ent) or enter a PDB ID to fetch from RCSB. Structures may include bound ligands if present in the PDB file. Supports batch processing of up to 10 structures. |
ProteinIQ supports a subset of safe native PROPKA options without changing default behavior:
chain:resnum residue pairsThe output includes a spreadsheet of detected ionizable groups plus a Files tab with two download types:
Those raw reports contain the full native text output, including the folding profile and charge profile sections that PROPKA writes after the summary table.
| Column | Description |
|---|---|
Structure | The source structure identifier, typically the fetched PDB ID or uploaded filename stem. |
Residue | Three-letter amino acid code (ASP, GLU, HIS, CYS, TYR, LYS, ARG, N+, or C-). |
Position | Residue number for protein groups, or atom label for ligand groups. |
Chain | Chain identifier from the PDB file. |
pKa | Predicted pKa value for this residue in the protein environment. |
Model pKa | Intrinsic pKa of the isolated amino acid in solution. |
Shift | Difference between predicted and model pKa (). |
Ligand atom type | native PROPKA ligand group/type annotation when the row comes from a ligand-derived titratable site. N/A means the row is a standard protein/terminal group with no ligand annotation. |
Coupling note | native note describing discarded or coupled alternative pKa values when applicable. N/A means PROPKA did not report a coupling note for that row. |
N/AProteinIQ now renders optional PROPKA fields as N/A instead of leaving them blank:
Ligand atom type = N/A — The row came from a standard protein residue or terminal group, not a ligand-derived titratable groupCoupling note = N/A — PROPKA did not attach a coupling/discard note to that rowN/A is intentional and means "not applicable here," not "missing due to a parser error."
propka-results.csv — Combined export of the normalized results table for all analyzed structures<structure>-propka-report.txt — Raw native PROPKA text report for each structureIf a field appears as N/A in the CSV, it has the same meaning as in the spreadsheet: the field was not applicable for that row.
PROPKA uses the following intrinsic pKa values as baselines:
| Residue | Model pKa |
|---|---|
| ASP (Aspartic acid) | 3.80 |
| GLU (Glutamic acid) | 4.50 |
| C-terminus | 3.20 |
| HIS (Histidine) | 6.50 |
| CYS (Cysteine) | 9.00 |
| TYR (Tyrosine) | 10.00 |
| LYS (Lysine) | 10.50 |
| ARG (Arginine) | 12.50 |
| N-terminus | 8.00 |
The Shift column indicates how the protein environment alters the intrinsic pKa:
To determine if a residue is protonated at a given pH:
At physiological pH (~7.4), residues with predicted pKa values near 7.4 may exist in mixed protonation states.
PROPKA computes pKa values by calculating a perturbation to the model (intrinsic) pKa caused by the protein environment. The predicted pKa is expressed as:
where DS represents desolvation effects, HB represents hydrogen bonding interactions, and CC represents Coulombic charge-charge interactions.
When an ionizable group is buried within a protein, it loses favorable solvation interactions with water. Desolvation destabilizes the charged form relative to the neutral form, shifting carboxylate pKa values upward (making them harder to deprotonate) and amine pKa values downward (making them harder to protonate).
PROPKA calculates the desolvation penalty from two factors:
The desolvation contribution scales continuously between surface and internal residues, avoiding the discontinuous behavior of earlier PROPKA versions that treated these as discrete categories.
Hydrogen bonds can stabilize either the charged or neutral form of an ionizable group, depending on the geometry and donor/acceptor relationship:
The hydrogen bonding contribution depends on both distance and angle, with optimal geometry producing the largest pKa shifts. PROPKA evaluates both side-chain and backbone hydrogen bond donors and acceptors.
Electrostatic interactions between ionizable groups affect their pKa values:
For buried residue pairs, PROPKA applies full Coulombic interactions. For surface-exposed pairs, the high dielectric of water screens these interactions substantially. The transition between these regimes uses the same smooth interpolation applied to desolvation effects.
When two ionizable groups interact strongly, their protonation states become coupled. Neither group can be assigned a single pKa value because the ionization of one depends on the protonation state of the other. PROPKA identifies these cases and marks them with an asterisk in detailed output.
Structures with missing atoms or poor resolution may produce unreliable results. Using PDB Fixer to repair structures before running PROPKA is recommended.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
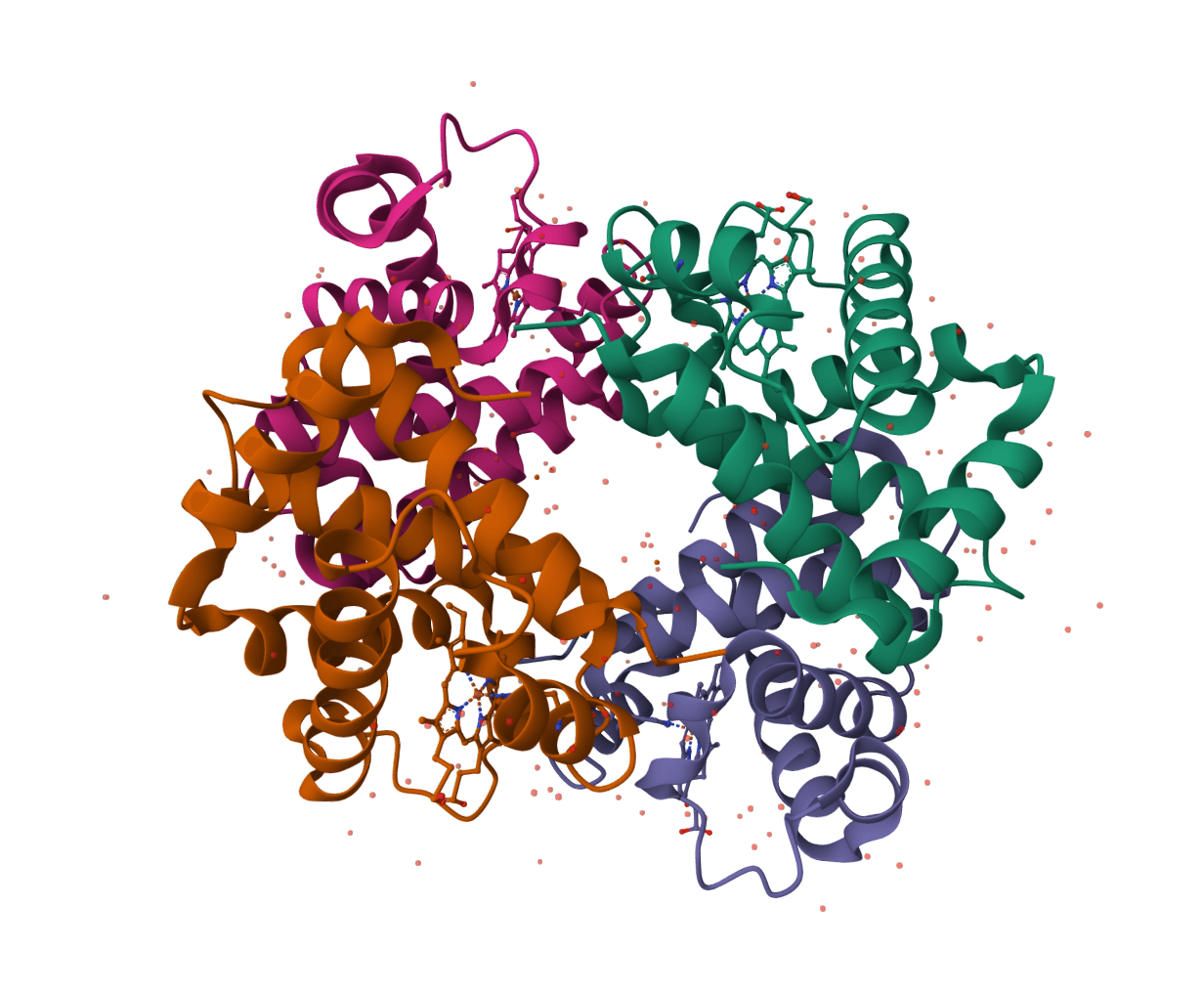
Compute 200+ RDKit molecular descriptors, drug-likeness rule violations, and structural fingerprints for QSAR, virtual screening, and ML workflows
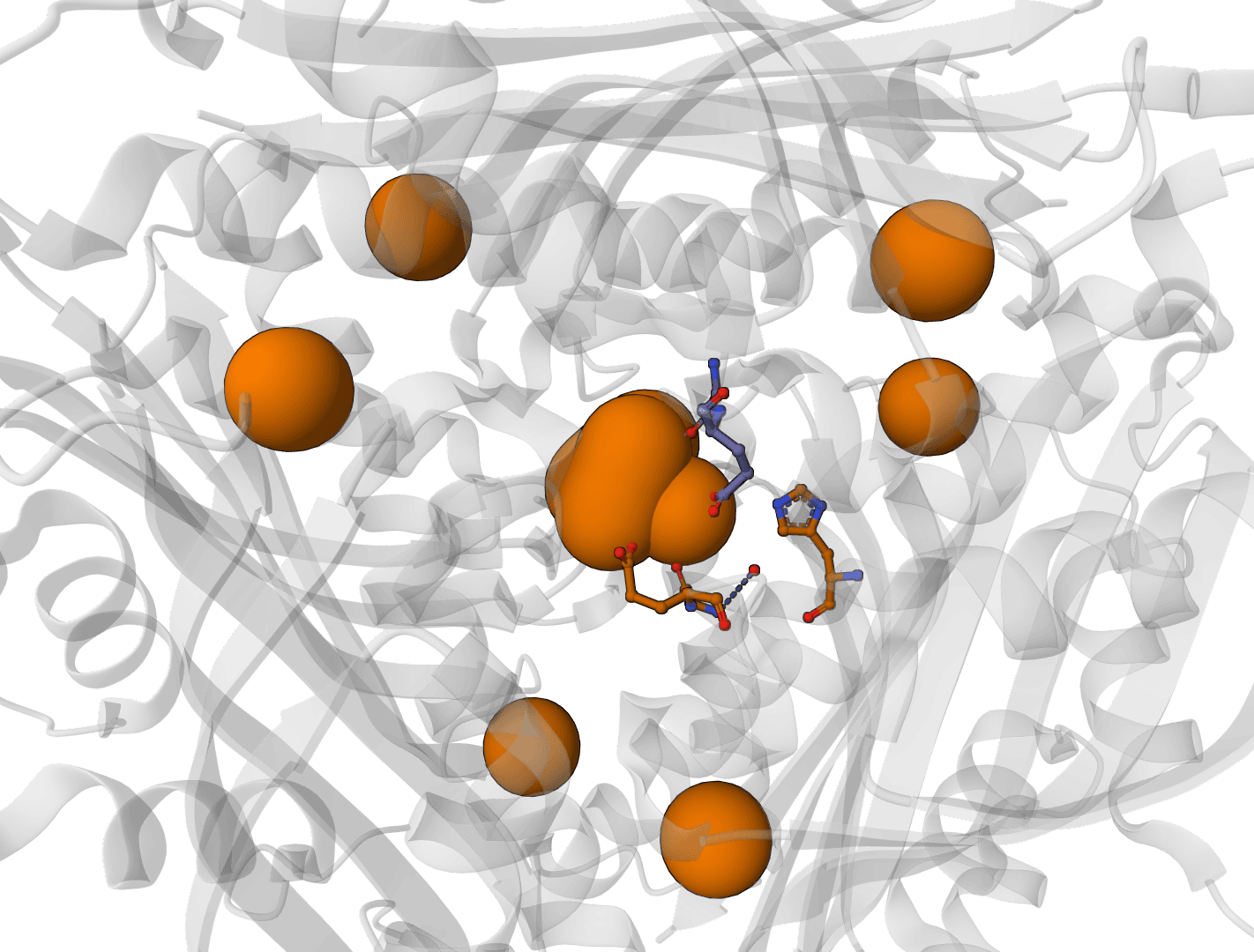
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Plot net charge vs pH for protein sequences. Visualize how protein charge changes across pH 0-14 and identify the isoelectric point (pI) where the net charge crosses zero.
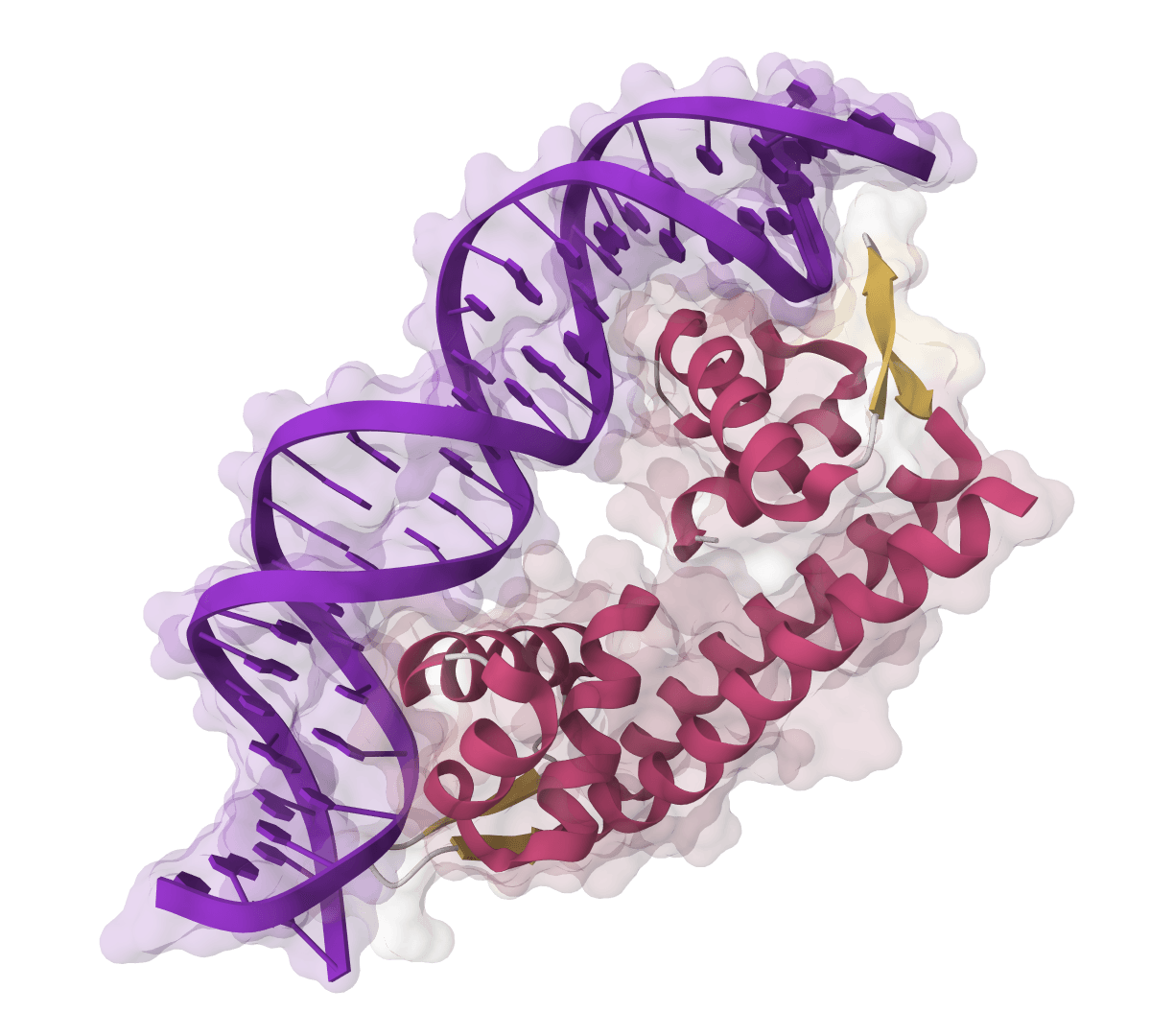
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Match experimental peptide masses against theoretical digest fragments of a protein sequence. Identify peptides from mass spectrometry data by peptide mass fingerprinting.
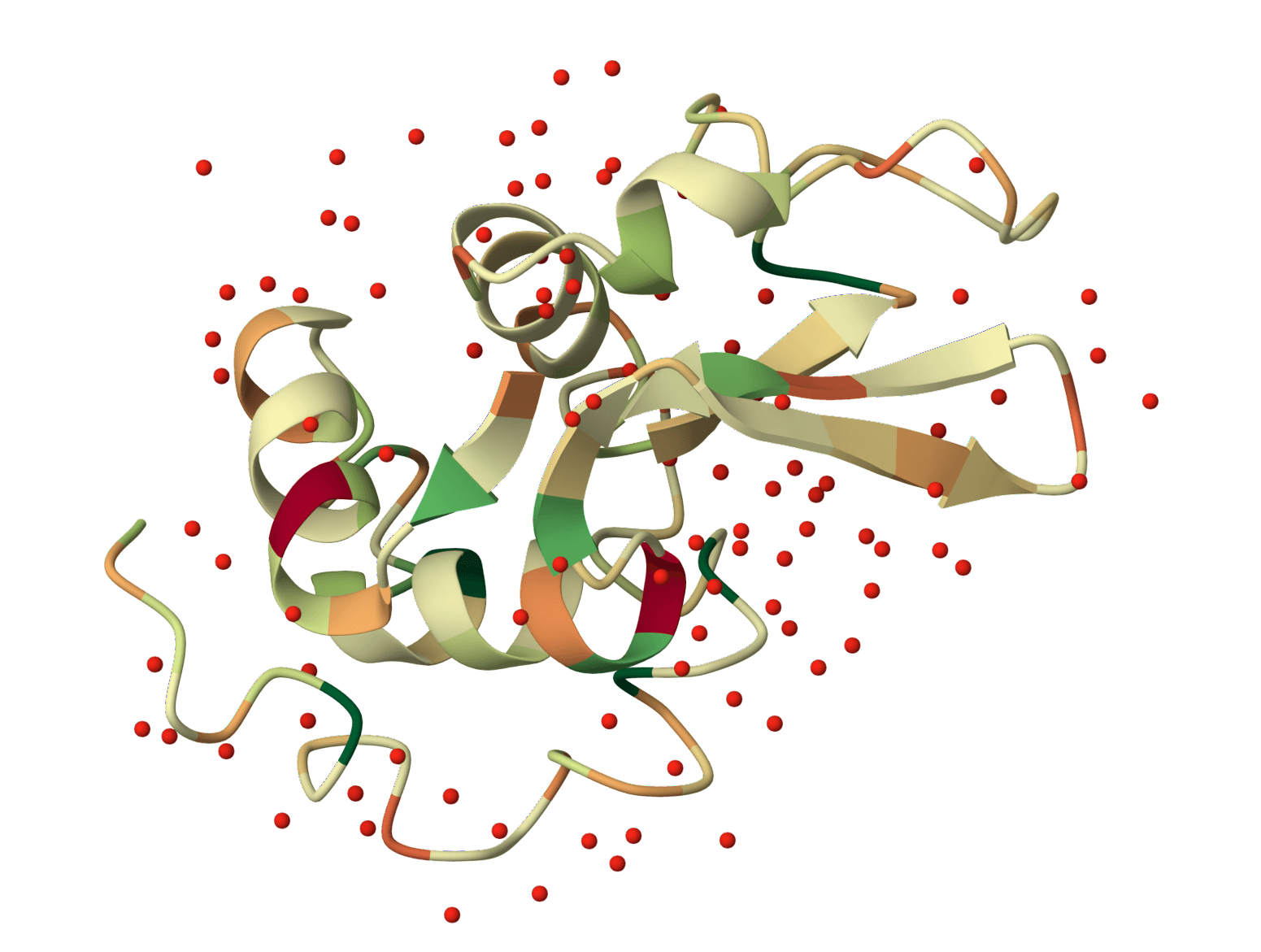
Generate Kyte-Doolittle hydropathy plots to visualize hydrophobic and hydrophilic regions along protein sequences. Identify transmembrane domains and surface-exposed regions.
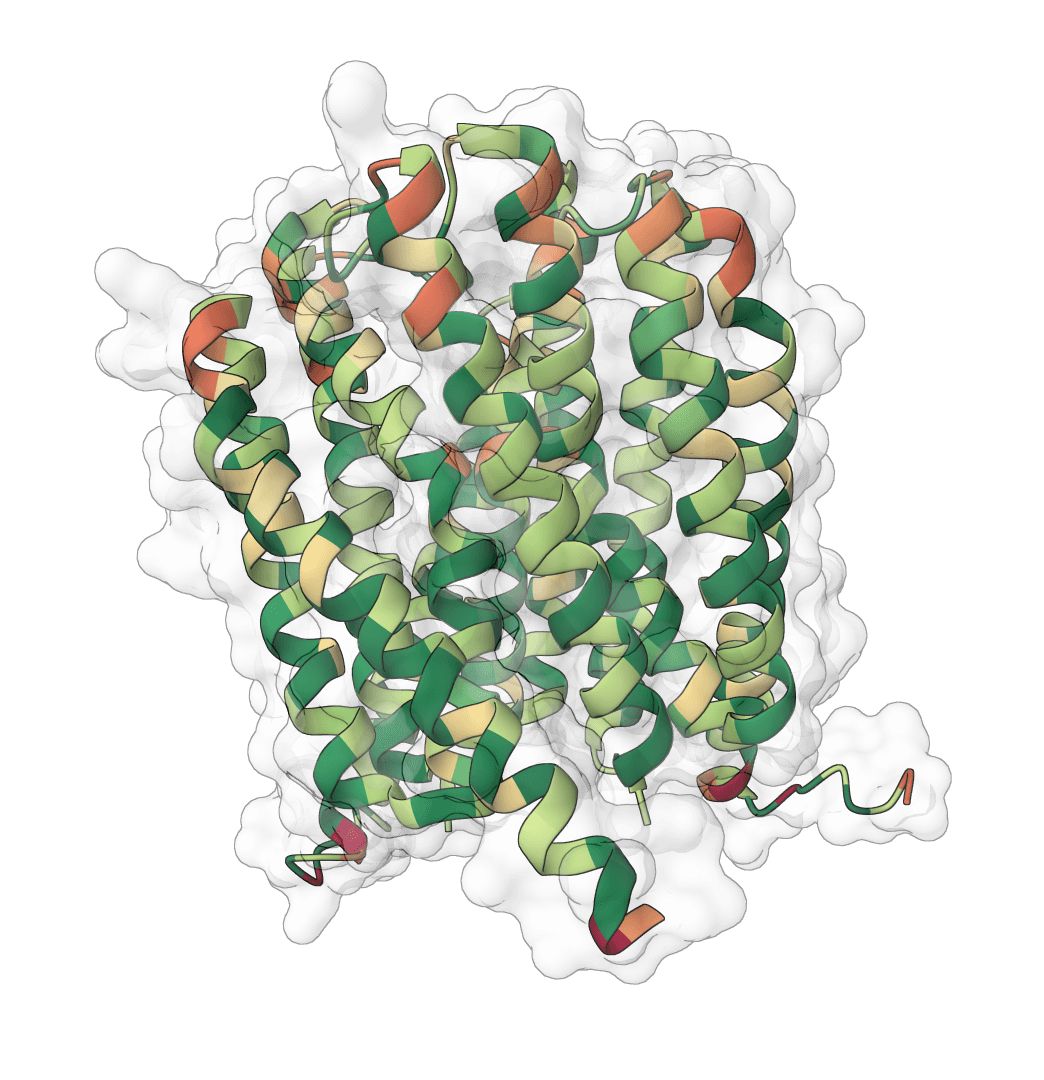
Generate hydrophobicity plots using 24 different amino acid scales. Visualize hydrophobic and hydrophilic regions for protein analysis, epitope prediction, and membrane protein studies.
Predict protease and chemical cleavage sites across a protein sequence for up to 39 enzymes simultaneously. Identify where each enzyme cuts, the cleavage residue, and context window around each site.
Cleave a protein sequence with a chosen protease and compute the masses of the resulting peptides. Supports multiple enzymes, missed cleavages, chemical modifications, and different ion types for mass spectrometry experiment planning.
PROPKA is an empirical method for predicting the pKa values of ionizable amino acid residues in proteins based on their three-dimensional structure. Developed by Jan H. Jensen and colleagues at the University of Copenhagen, PROPKA calculates how the protein environment shifts the pKa of each ionizable group away from its intrinsic (model) value.
The pKa of an ionizable group determines at what pH that group gains or loses a proton. In solution, amino acids have characteristic pKa values, but within a folded protein, factors such as burial from solvent, hydrogen bonding, and electrostatic interactions with nearby charged residues can shift these values substantially. PROPKA quantifies these perturbations using structure-based empirical energy functions, enabling rapid prediction of protonation states at any given pH.
PROPKA 3 introduced consistent treatment of internal and surface residues through improved desolvation and dielectric response modeling. The method handles ASP, GLU, HIS, CYS, TYR, LYS, ARG residues and both N- and C-terminal groups. Benchmarks show root-mean-square deviations of 0.79 pKa units for Asp/Glu, 0.75 for Tyr, 0.65 for Lys, and 1.00 for His residues compared to experimental measurements.
ProteinIQ provides a web-based interface for running PROPKA 3.5.1 without command-line installation. Upload a protein structure file or fetch one from RCSB, and receive predicted pKa values for ionizable residues and supported ligand groups present in the structure.
| Input | Description |
|---|---|
Protein Structures | The target protein structure. Upload a PDB file (.pdb or .ent) or enter a PDB ID to fetch from RCSB. Structures may include bound ligands if present in the PDB file. Supports batch processing of up to 10 structures. |
ProteinIQ supports a subset of safe native PROPKA options without changing default behavior:
chain:resnum residue pairsThe output includes a spreadsheet of detected ionizable groups plus a Files tab with two download types:
Those raw reports contain the full native text output, including the folding profile and charge profile sections that PROPKA writes after the summary table.
| Column | Description |
|---|---|
Structure | The source structure identifier, typically the fetched PDB ID or uploaded filename stem. |
Residue | Three-letter amino acid code (ASP, GLU, HIS, CYS, TYR, LYS, ARG, N+, or C-). |
Position | Residue number for protein groups, or atom label for ligand groups. |
Chain | Chain identifier from the PDB file. |
pKa | Predicted pKa value for this residue in the protein environment. |
Model pKa | Intrinsic pKa of the isolated amino acid in solution. |
Shift | Difference between predicted and model pKa (). |
Ligand atom type | native PROPKA ligand group/type annotation when the row comes from a ligand-derived titratable site. N/A means the row is a standard protein/terminal group with no ligand annotation. |
Coupling note | native note describing discarded or coupled alternative pKa values when applicable. N/A means PROPKA did not report a coupling note for that row. |
N/AProteinIQ now renders optional PROPKA fields as N/A instead of leaving them blank:
Ligand atom type = N/A — The row came from a standard protein residue or terminal group, not a ligand-derived titratable groupCoupling note = N/A — PROPKA did not attach a coupling/discard note to that rowN/A is intentional and means "not applicable here," not "missing due to a parser error."
propka-results.csv — Combined export of the normalized results table for all analyzed structures<structure>-propka-report.txt — Raw native PROPKA text report for each structureIf a field appears as N/A in the CSV, it has the same meaning as in the spreadsheet: the field was not applicable for that row.
PROPKA uses the following intrinsic pKa values as baselines:
| Residue | Model pKa |
|---|---|
| ASP (Aspartic acid) | 3.80 |
| GLU (Glutamic acid) | 4.50 |
| C-terminus | 3.20 |
| HIS (Histidine) | 6.50 |
| CYS (Cysteine) | 9.00 |
| TYR (Tyrosine) | 10.00 |
| LYS (Lysine) | 10.50 |
| ARG (Arginine) | 12.50 |
| N-terminus | 8.00 |
The Shift column indicates how the protein environment alters the intrinsic pKa:
To determine if a residue is protonated at a given pH:
At physiological pH (~7.4), residues with predicted pKa values near 7.4 may exist in mixed protonation states.
PROPKA computes pKa values by calculating a perturbation to the model (intrinsic) pKa caused by the protein environment. The predicted pKa is expressed as:
where DS represents desolvation effects, HB represents hydrogen bonding interactions, and CC represents Coulombic charge-charge interactions.
When an ionizable group is buried within a protein, it loses favorable solvation interactions with water. Desolvation destabilizes the charged form relative to the neutral form, shifting carboxylate pKa values upward (making them harder to deprotonate) and amine pKa values downward (making them harder to protonate).
PROPKA calculates the desolvation penalty from two factors:
The desolvation contribution scales continuously between surface and internal residues, avoiding the discontinuous behavior of earlier PROPKA versions that treated these as discrete categories.
Hydrogen bonds can stabilize either the charged or neutral form of an ionizable group, depending on the geometry and donor/acceptor relationship:
The hydrogen bonding contribution depends on both distance and angle, with optimal geometry producing the largest pKa shifts. PROPKA evaluates both side-chain and backbone hydrogen bond donors and acceptors.
Electrostatic interactions between ionizable groups affect their pKa values:
For buried residue pairs, PROPKA applies full Coulombic interactions. For surface-exposed pairs, the high dielectric of water screens these interactions substantially. The transition between these regimes uses the same smooth interpolation applied to desolvation effects.
When two ionizable groups interact strongly, their protonation states become coupled. Neither group can be assigned a single pKa value because the ionization of one depends on the protonation state of the other. PROPKA identifies these cases and marks them with an asterisk in detailed output.
Structures with missing atoms or poor resolution may produce unreliable results. Using PDB Fixer to repair structures before running PROPKA is recommended.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Compute 200+ RDKit molecular descriptors, drug-likeness rule violations, and structural fingerprints for QSAR, virtual screening, and ML workflows
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Plot net charge vs pH for protein sequences. Visualize how protein charge changes across pH 0-14 and identify the isoelectric point (pI) where the net charge crosses zero.
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Match experimental peptide masses against theoretical digest fragments of a protein sequence. Identify peptides from mass spectrometry data by peptide mass fingerprinting.
Generate Kyte-Doolittle hydropathy plots to visualize hydrophobic and hydrophilic regions along protein sequences. Identify transmembrane domains and surface-exposed regions.
Generate hydrophobicity plots using 24 different amino acid scales. Visualize hydrophobic and hydrophilic regions for protein analysis, epitope prediction, and membrane protein studies.
Predict protease and chemical cleavage sites across a protein sequence for up to 39 enzymes simultaneously. Identify where each enzyme cuts, the cleavage residue, and context window around each site.
Cleave a protein sequence with a chosen protease and compute the masses of the resulting peptides. Supports multiple enzymes, missed cleavages, chemical modifications, and different ion types for mass spectrometry experiment planning.