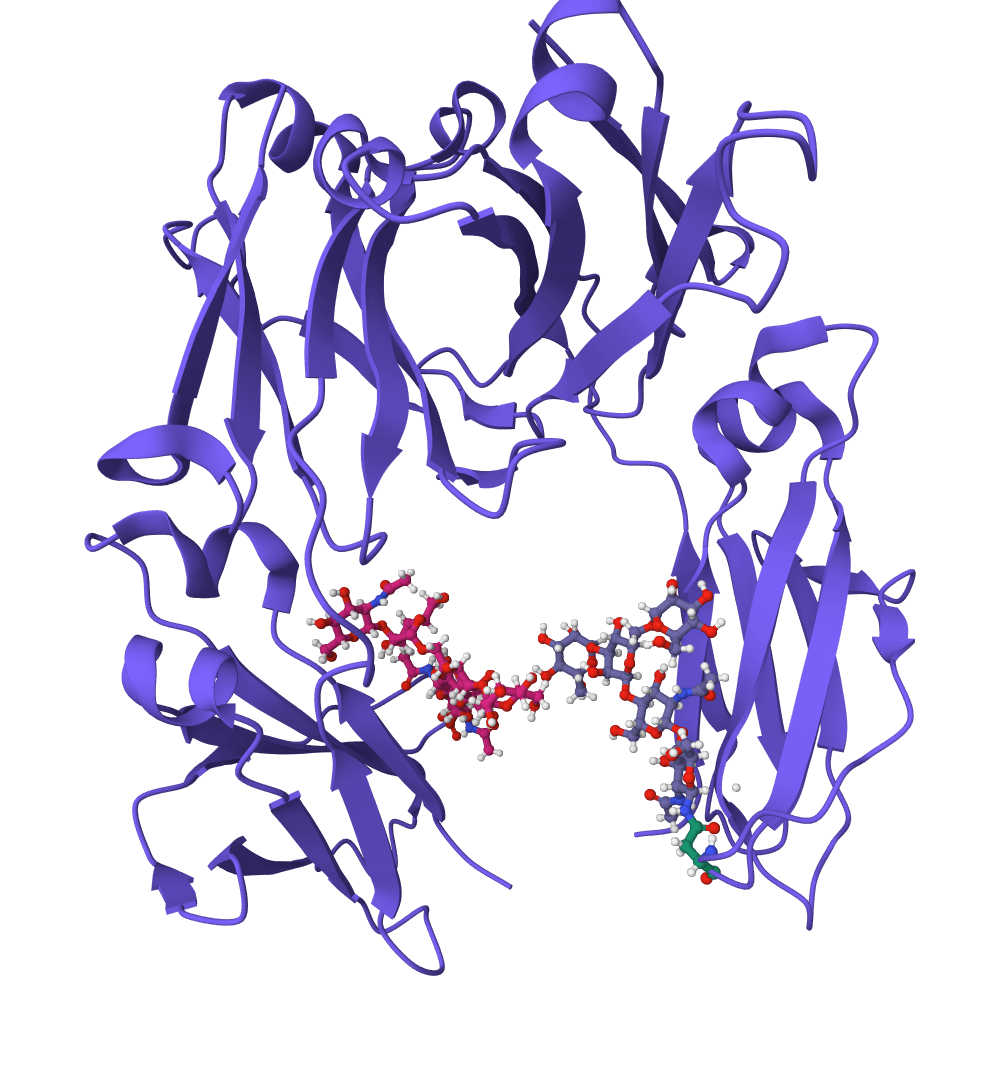ScanNet (Spatio-Chemical Arrangement of Neighbors Network) is a geometric deep learning model for predicting protein binding sites directly from 3D structures. It identifies where proteins interact with other proteins, antibodies, or intrinsically disordered proteins (IDPs).
Unlike traditional methods that rely on handcrafted features or structural homology, ScanNet learns spatio-chemical patterns end-to-end from atomic coordinates. The model constructs representations by examining the spatial and chemical arrangement of neighboring atoms, enabling it to detect binding sites even on novel protein folds not seen during training.
Published in Nature Methods in 2022, ScanNet demonstrated state-of-the-art accuracy on multiple benchmarks while remaining interpretable through visualization of learned filters.
Recommended ScanNet applications:
Known ScanNet limitations:
ProteinIQ provides a web interface for running ScanNet without command-line installation or local database setup. Upload a protein structure, select a prediction mode, and receive per-residue binding probabilities.
| Input | Description |
|---|---|
Protein Structure | The target protein to analyze. Upload a PDB or mmCIF file, enter a PDB ID (e.g., 1brs) for RCSB structures, or enter a UniProt ID (e.g., P38398) for AlphaFold structures. |
| Setting | Description |
|---|---|
Prediction mode | Type of binding site to predict. General binding sites (default) detects protein-protein interfaces. Antibody epitopes identifies B-cell epitope regions. IDP binding sites locates interaction sites for disordered proteins. |
Chain selection | Specific chain(s) to analyze (e.g., A or AB). Leave empty to analyze all chains. For PDB IDs, underscore notation also works in the input field (e.g., 1brs_A). |
Process chains together | Runs ScanNet assembly mode, processing selected chains jointly instead of independently. Off by default to preserve ScanNet's standard behavior. |
ProteinIQ runs ScanNet in no-MSA mode for this online job type, so predictions use the submitted structure without building multiple sequence alignments.
ScanNet returns a table of per-residue binding site predictions and downloadable native result files, including the prediction CSV and annotated structure or visualization scripts when ScanNet writes them.
| Column | Description |
|---|---|
Residue | Position in the protein sequence. |
Chain | Chain identifier from the input structure. |
AA | Single-letter amino acid code at this position. |
Probability | Predicted likelihood (0–1) that this residue is part of a binding interface. |
Higher probabilities indicate residues ScanNet ranks as more likely to participate in a binding interface. Use the per-residue values together with the annotated structure outputs to inspect contiguous surface patches rather than relying on a single residue in isolation. ScanNet does not define universal high/medium/low probability cutoffs, so interpret the score distribution in the context of the submitted structure.
ScanNet employs a hierarchical architecture that processes protein structures at both atomic and amino acid scales, learning to recognize spatio-chemical patterns associated with binding interfaces.
At the core of ScanNet are trainable filters that detect specific spatial arrangements of atoms with particular chemical properties. For each atom in the structure, neighboring atoms within a local coordinate frame are extracted. These point clouds pass through linear filters that respond to arrangements such as "hydrophobic atoms surrounding a polar center" or "aromatic ring flanked by charged residues."
The filters are parameterized to be interpretable—each can be visualized to understand what molecular pattern it detects. This interpretability distinguishes ScanNet from black-box deep learning approaches.
ScanNet builds representations at two scales:
This multi-scale approach allows ScanNet to integrate information from individual atomic contacts up to residue-level surface geometry.
When MSA data is available in a configured ScanNet installation, the method can incorporate position-weight matrices derived from sequence alignments. Conservation patterns provide complementary evidence because residues conserved across evolution often participate in functionally important interfaces. The ProteinIQ online job runs the no-MSA mode, which relies on structural features.
ScanNet was trained on protein-protein binding sites from the MaSIF-site benchmark dataset. Testing revealed strong generalization: even for proteins with no sequence or fold similarity to training examples, ScanNet maintained high accuracy. This contrasts with homology-based methods that fail on novel folds.
ScanNet achieves state-of-the-art accuracy on protein-protein binding site prediction benchmarks.
| Metric | Value |
|---|---|
| AUCPR (test set) | 0.694 |
| Accuracy | 87.7% |
| Precision at 50% recall | 73.5% |
Performance remains robust across different levels of similarity to training data. While structural homology methods excel when close homologs exist, their accuracy degrades rapidly for distant or novel folds. ScanNet maintains consistent performance across all homology levels, demonstrating true generalization rather than memorization.
Predictions are also robust to conformational changes between bound and unbound structures, with only minor accuracy drops (88.3% to 86.6% on simulated data, 91.9% to 91.3% on experimental structures).
Analyze noncovalent interactions in protein-ligand complex structures with PLIP, including hydrogen bonds, hydrophobic contacts, pi interactions, salt bridges, water bridges, halogen bonds, and metal complexes.
ProLIF calculates protein-ligand interaction fingerprints from 3D structures, returning residue-level interaction tables, interaction metadata, and native fingerprint files.
Restore missing residues in antibody sequences using a language model trained on the Observed Antibody Space (OAS) database. Achieves better restoration than IMGT germlines or ESM-1b while being 7x faster.
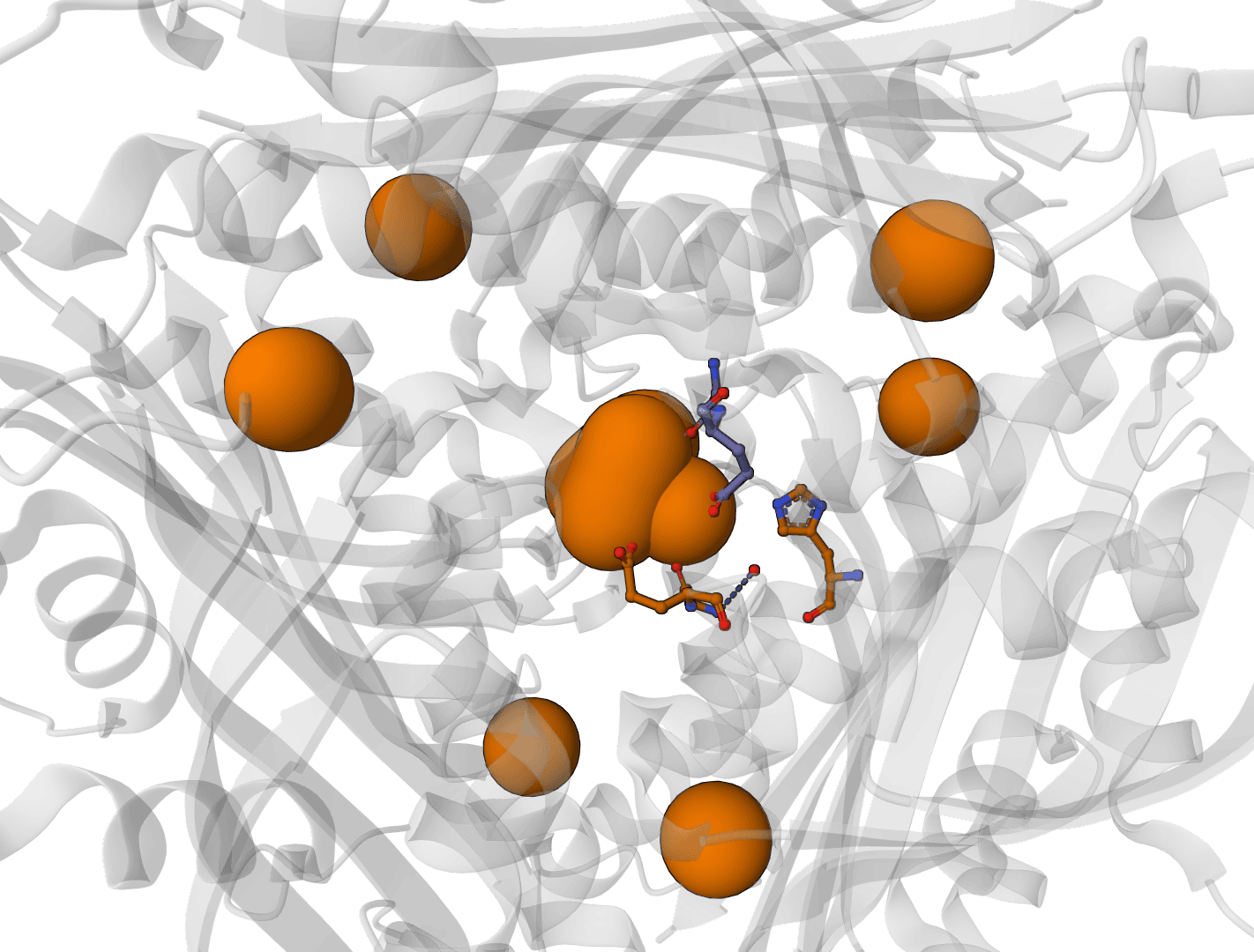
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
LocScale performs physics-informed local sharpening of cryo-EM density maps using half-maps or full MRC/MAP volumes, with optional mask and reference-map inputs.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
ScanNet (Spatio-Chemical Arrangement of Neighbors Network) is a geometric deep learning model for predicting protein binding sites directly from 3D structures. It identifies where proteins interact with other proteins, antibodies, or intrinsically disordered proteins (IDPs).
Unlike traditional methods that rely on handcrafted features or structural homology, ScanNet learns spatio-chemical patterns end-to-end from atomic coordinates. The model constructs representations by examining the spatial and chemical arrangement of neighboring atoms, enabling it to detect binding sites even on novel protein folds not seen during training.
Published in Nature Methods in 2022, ScanNet demonstrated state-of-the-art accuracy on multiple benchmarks while remaining interpretable through visualization of learned filters.
Recommended ScanNet applications:
Known ScanNet limitations:
ProteinIQ provides a web interface for running ScanNet without command-line installation or local database setup. Upload a protein structure, select a prediction mode, and receive per-residue binding probabilities.
| Input | Description |
|---|---|
Protein Structure | The target protein to analyze. Upload a PDB or mmCIF file, enter a PDB ID (e.g., 1brs) for RCSB structures, or enter a UniProt ID (e.g., P38398) for AlphaFold structures. |
| Setting | Description |
|---|---|
Prediction mode | Type of binding site to predict. General binding sites (default) detects protein-protein interfaces. Antibody epitopes identifies B-cell epitope regions. IDP binding sites locates interaction sites for disordered proteins. |
Chain selection | Specific chain(s) to analyze (e.g., A or AB). Leave empty to analyze all chains. For PDB IDs, underscore notation also works in the input field (e.g., 1brs_A). |
Process chains together | Runs ScanNet assembly mode, processing selected chains jointly instead of independently. Off by default to preserve ScanNet's standard behavior. |
ProteinIQ runs ScanNet in no-MSA mode for this online job type, so predictions use the submitted structure without building multiple sequence alignments.
ScanNet returns a table of per-residue binding site predictions and downloadable native result files, including the prediction CSV and annotated structure or visualization scripts when ScanNet writes them.
| Column | Description |
|---|---|
Residue | Position in the protein sequence. |
Chain | Chain identifier from the input structure. |
AA | Single-letter amino acid code at this position. |
Probability | Predicted likelihood (0–1) that this residue is part of a binding interface. |
Higher probabilities indicate residues ScanNet ranks as more likely to participate in a binding interface. Use the per-residue values together with the annotated structure outputs to inspect contiguous surface patches rather than relying on a single residue in isolation. ScanNet does not define universal high/medium/low probability cutoffs, so interpret the score distribution in the context of the submitted structure.
ScanNet employs a hierarchical architecture that processes protein structures at both atomic and amino acid scales, learning to recognize spatio-chemical patterns associated with binding interfaces.
At the core of ScanNet are trainable filters that detect specific spatial arrangements of atoms with particular chemical properties. For each atom in the structure, neighboring atoms within a local coordinate frame are extracted. These point clouds pass through linear filters that respond to arrangements such as "hydrophobic atoms surrounding a polar center" or "aromatic ring flanked by charged residues."
The filters are parameterized to be interpretable—each can be visualized to understand what molecular pattern it detects. This interpretability distinguishes ScanNet from black-box deep learning approaches.
ScanNet builds representations at two scales:
This multi-scale approach allows ScanNet to integrate information from individual atomic contacts up to residue-level surface geometry.
When MSA data is available in a configured ScanNet installation, the method can incorporate position-weight matrices derived from sequence alignments. Conservation patterns provide complementary evidence because residues conserved across evolution often participate in functionally important interfaces. The ProteinIQ online job runs the no-MSA mode, which relies on structural features.
ScanNet was trained on protein-protein binding sites from the MaSIF-site benchmark dataset. Testing revealed strong generalization: even for proteins with no sequence or fold similarity to training examples, ScanNet maintained high accuracy. This contrasts with homology-based methods that fail on novel folds.
ScanNet achieves state-of-the-art accuracy on protein-protein binding site prediction benchmarks.
| Metric | Value |
|---|---|
| AUCPR (test set) | 0.694 |
| Accuracy | 87.7% |
| Precision at 50% recall | 73.5% |
Performance remains robust across different levels of similarity to training data. While structural homology methods excel when close homologs exist, their accuracy degrades rapidly for distant or novel folds. ScanNet maintains consistent performance across all homology levels, demonstrating true generalization rather than memorization.
Predictions are also robust to conformational changes between bound and unbound structures, with only minor accuracy drops (88.3% to 86.6% on simulated data, 91.9% to 91.3% on experimental structures).
Analyze noncovalent interactions in protein-ligand complex structures with PLIP, including hydrogen bonds, hydrophobic contacts, pi interactions, salt bridges, water bridges, halogen bonds, and metal complexes.
ProLIF calculates protein-ligand interaction fingerprints from 3D structures, returning residue-level interaction tables, interaction metadata, and native fingerprint files.
Restore missing residues in antibody sequences using a language model trained on the Observed Antibody Space (OAS) database. Achieves better restoration than IMGT germlines or ESM-1b while being 7x faster.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
LocScale performs physics-informed local sharpening of cryo-EM density maps using half-maps or full MRC/MAP volumes, with optional mask and reference-map inputs.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.