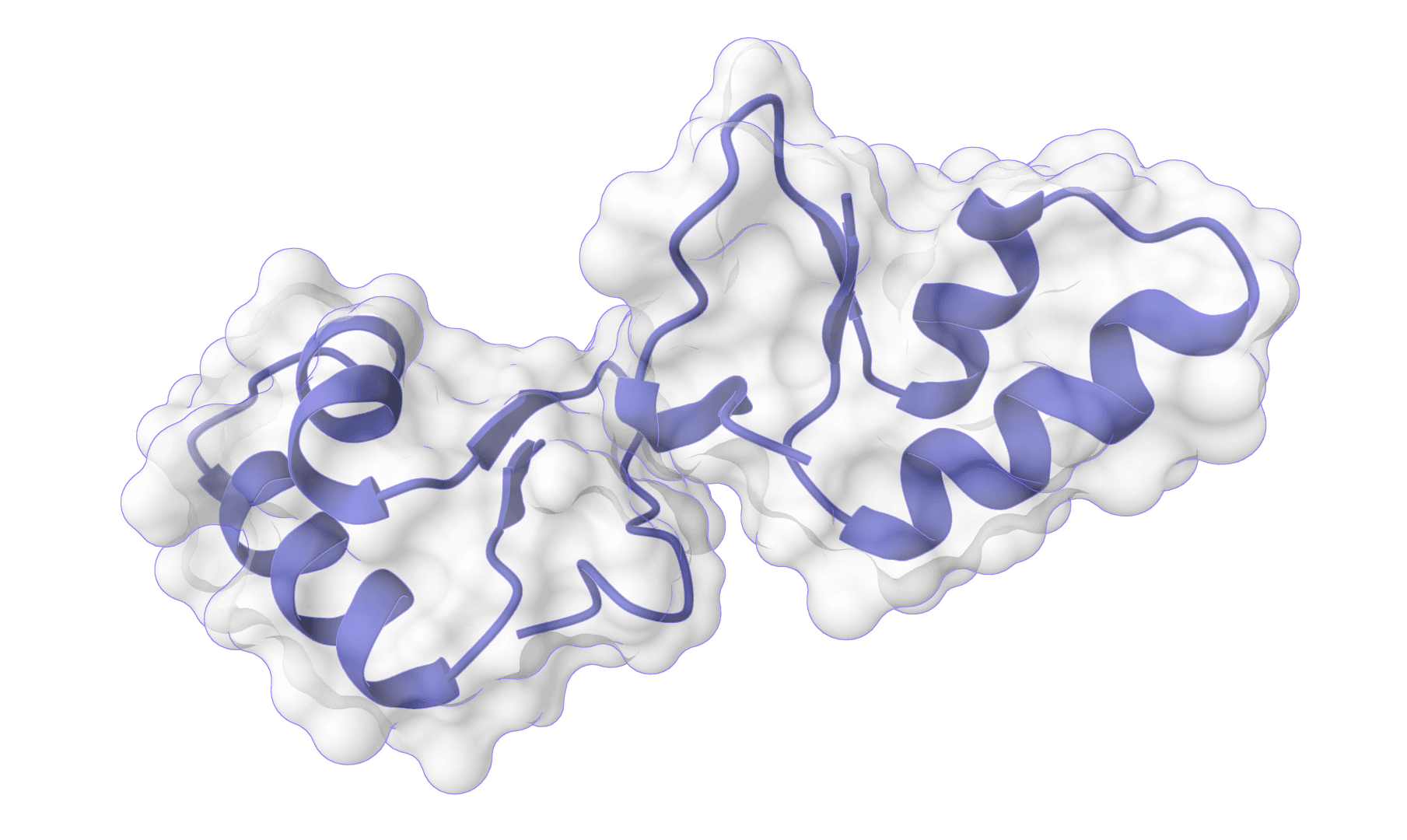
Integrative protein-protein docking guided by experimental restraints Learn more
Run
50 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
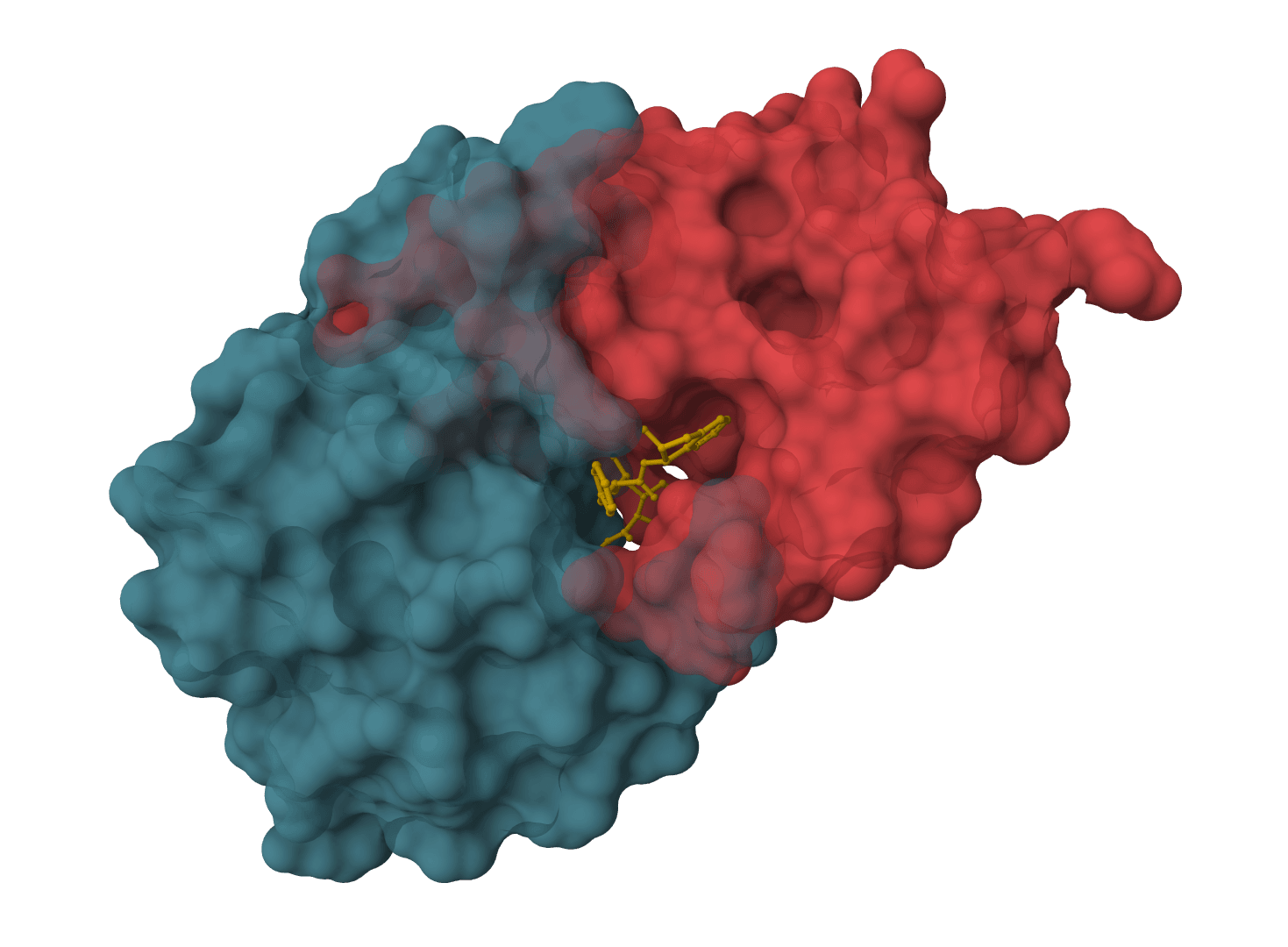
Influenza HA nanobody redocking
Integrative protein-protein docking guided by experimental restraints Learn more
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Influenza HA nanobody redocking
Integrative protein-protein docking guided by experimental restraints Learn more
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Influenza HA nanobody redocking
HADDOCK3 (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for predicting the structure of biomolecular complexes. Unlike docking methods that rely solely on shape complementarity, HADDOCK3 incorporates experimental or bioinformatic data as restraints to guide the search. This makes it particularly effective when partial interface information is available from mutagenesis, NMR chemical shift perturbations, cross-linking mass spectrometry, or epitope mapping.
HADDOCK3 is developed by the Bonvin Lab at Utrecht University and represents a complete redesign of the original HADDOCK platform with a fully modular workflow.
Upload two protein structures (PDB or mmCIF format, or enter a PDB ID) as the receptor and ligand, optionally provide interface residue numbers for each partner, and submit the job. ProteinIQ returns ranked docked complexes, the complete per-model and per-cluster CAPRI tables, source energy terms, clustering files, the effective configuration, the run log, and a bounded native analysis archive.
| Input | Format | Notes |
|---|---|---|
Receptor | PDB, ENT, CIF, mmCIF, or PDB ID | Protein atoms required. mmCIF inputs are converted to PDB automatically. |
Ligand | PDB, ENT, CIF, mmCIF, or PDB ID | The second docking partner, typically the smaller protein. |
Active residues | Comma-separated numbers or ranges (e.g., 38,40,45 or 38-45) | Residues directly involved at the interface. |
Passive residues | Same format | Surface residues near active residues that may contribute to binding. |
Chain ID | Single letter | Leave blank to use the first detected chain after preprocessing. |
| Setting | Default | Description |
|---|---|---|
Number of models | 50 | Rigid-body sampling count. Start with 50; use 100-200 with reliable restraints. |
Top models to return | 10 | Final complexes returned after clustering and refinement. |
Ab initio mode | Center-of-mass | Fallback guidance when no restraints are provided. cmrest applies center-of-mass restraints; ranair uses random AIRs; none docks freely. |
CPU cores | 8 | Parallelization used by HADDOCK3 inside the 8-core runtime. |
Rigid body tolerance | 5 | Percentage of failed rigidbody models accepted before the run aborts (default). |
Flexref tolerance | 5 | Same as above for the semi-flexible refinement stage. |
EM refinement tolerance | 5 | Same for energy minimization. |
Select top models before refinement | Automatic | Uses the HADDOCK3 default of 200, capped at the rigid-body sampling count. |
Minimum cluster population | 1 | Clusters with fewer members than this are discarded. Set low for small sampling counts. |
Models retained per cluster | 10 | Models retained from each selected cluster for CAPRI evaluation. This is separate from the total number returned. |
Each returned complex includes its HADDOCK score, native energy terms, cluster identity, and CAPRI fields. Because the current form does not accept an experimental reference complex, RMSD, Fnat, and DockQ values are relative to HADDOCK3's lowest-scoring run model, not an experimentally determined native structure.
| Column | Description |
|---|---|
rank | Global rank by HADDOCK score (lower is better). |
score | Weighted HADDOCK score in arbitrary units. |
vdw | Intermolecular van der Waals energy (kcal/mol). |
elec | Intermolecular electrostatic energy (kcal/mol). |
desolv | Empirical desolvation energy (kcal/mol). |
air | Ambiguous interaction restraint violation energy (kcal/mol). |
bsa | Buried surface area at the interface (Ų). |
cluster | Cluster identifier. Models in the same cluster adopt similar interface conformations. |
dockq, irmsd, fnat, lrmsd, ilrmsd | CAPRI comparison metrics relative to the lowest-scoring run model. |
total, angles, bonds, cdih, coup, dani, dihe, improper, rdcs, rg, vean, xpcs | Additional native energy and structural terms returned by CAPRI evaluation. |
The Files tab also includes the untouched capri_ss.tsv, capri_clt.tsv, FCC clustering tables, effective workflow configuration, run log, bounded analysis-report archive, and an artifact manifest. Coordinate and native-artifact payloads use aggregate limits; files omitted from an unusually large result are listed explicitly instead of being allowed to overload completion processing.
The core concept is the Ambiguous Interaction Restraint. An AIR encodes experimental uncertainty without requiring exact atomic contacts: it defines a distance restraint between any atom of an active residue on one protein and any atom on the active or passive residues of the partner. This flexibility accommodates the fact that most experimental techniques identify interface-proximal residues, not specific atomic contacts.
Active residues incur an energy penalty if they remain solvent-exposed in the docked complex. Passive residues are allowed but not required to participate. Random removal of a fraction of restraints during each docking trial ensures the search samples a range of conformations even when some restraints are incorrect.
The ProteinIQ implementation runs a five-stage pipeline:
Stage 1 - Topology generation (topoaa): Generates CNS-format all-atom topology for both proteins. Histidine protonation states are assigned automatically.
Stage 2 - Rigid body docking (rigidbody): Proteins are randomized in orientation and subjected to rigid-body energy minimization guided by AIRs or ab-initio restraints. The Number of models setting controls how many structures are generated here.
Stage 3 - Semi-flexible refinement (flexref): Top-scoring rigid-body models undergo simulated annealing in torsion angle space. Side chains and backbone atoms near the interface are free to move, optimizing local contacts without perturbing the global fold.
Stage 4 - Energy minimization refinement (emref): Selected models are refined by Cartesian energy minimization with electrostatics, van der Waals forces, and desolvation terms. This produces the final scored coordinates.
Stage 5 - Clustering and evaluation (clustfcc, seletopclusts, caprieval): Models are grouped by fraction of common contacts (FCC). Top models per cluster are selected, then scored by CAPRI evaluation, which computes the per-model energy breakdown and global ranking used in the results table.
The final HADDOCK score is a weighted sum of energy terms evaluated at the emref stage:
The heavy weight on van der Waals and desolvation reflects their central role in protein-protein recognition. Electrostatics is downweighted because its long-range character and sensitivity to dielectric treatment make it less reliable as a discriminator. Individual term values are returned alongside the composite score so that models can be compared by specific interaction type.
Typical scores for crystallographically validated complexes fall between -100 and -200. Absolute values scale with interface size, so ranking within a run is more informative than comparing scores across different protein pairs.
Both receptor and ligand accept multi-chain PDB files. A pinned-version compatibility reproducer showed that HADDOCK3's rigidbody CNS script can index more chain segments than docking partners, causing an undefined mol_fix_origin_N variable.
ProteinIQ retains the narrow compatibility workaround by collapsing each upload into a single docking body before the maintained HADDOCK3 preprocessor runs. Chain A is used for the receptor body and chain B for the ligand body. If interface residues are specified per chain, residue numbers are remapped onto the collapsed structure before source restraint generation. Multi-model PDB ensemble boundaries are preserved. When no restraints are provided for a multi-chain input, center-of-mass restraints (cmrest) are enabled automatically to maintain intermolecular guidance during rigid-body docking.
For single-chain inputs, overlapping chain IDs between receptor and ligand are remapped automatically (for example, if both use chain A, the ligand is reassigned to chain C before docking).
The vdw term is usually the most reliable discriminator between good and poor models. A very positive value (above +50) suggests steric clashes that were not resolved during refinement. The desolv term captures burial of hydrophobic surface; large negative values indicate a well-packed hydrophobic core. An air value significantly above zero means active residues are not forming the expected contacts, which can indicate incorrect restraints or a poorly sampled binding mode.
The buried surface area (bsa) is not part of the score formula but correlates with interface size. Values below 500 Ų typically indicate poor packing; values above 1500 Ų suggest large, well-formed interfaces.
Clusters with high population and low average score represent binding modes that are both energetically favorable and structurally consistent across multiple independent trials. When the top cluster contains most models, the docking has converged on a single binding mode. Fragmented clusters with similar scores suggest an underdefined interface, where increasing sampling or adding restraints would help.
If restraints are reliable, the top-ranked model within the largest cluster is most likely to approximate the native binding mode. For completely unknown interfaces (ab initio mode), the top 2-3 clusters should be examined since multiple modes may be plausible.
Data-driven docking uses active and passive residues to focus the search on known interface regions. Success rates exceed 70% in benchmark studies when restraints are correctly assigned. Convergence is faster because the rigid-body sampling is not exploring the full translational and rotational space.
Ab initio docking runs when no restraints are provided. HADDOCK3 generates random AIRs automatically (ranair), or restricts sampling using center-of-mass restraints (cmrest). Success rates drop to 30-40%, but the mode is useful when no interface information is available or when testing whether experimental observations are consistent with a proposed binding site.
Center-of-mass restraints (cmrest, the default) represent the best trade-off for ab initio runs: they ensure the proteins come into proximity without biasing which surface forms the interface.
HADDOCK3 is the right choice when some experimental interface information is available. The data-driven mode consistently outperforms purely computational approaches for these cases. It handles protein-protein complexes of arbitrary size, including antibody-antigen, enzyme-substrate, and homo-oligomeric systems.
LightDock is faster for exploratory runs with no interface data, using swarm intelligence to sample a wide range of docking poses. It does not support AIRs natively, so it loses the advantage HADDOCK3 has when restraints are available.
ColabDock and EquiDock are deep learning approaches that require no restraints and run faster, but cannot incorporate experimental data the way HADDOCK3 can.
For protein-ligand docking (small molecule into a protein binding site), use AutoDock Vina, GNINA, or DiffDock instead. HADDOCK3 is designed for macromolecular interfaces.
NMR-based complex modeling: Chemical shift perturbations identify residues affected by binding. Map perturbed residues to active, surface-adjacent residues to passive, and submit. HADDOCK3's accuracy in this regime is well established across hundreds of benchmark complexes.
Mutagenesis-guided docking: Mutations that disrupt binding localize the interface. Alanine scan results map directly to active residues. Surrounding exposed surface residues serve as passive restraints.
Antibody-antigen docking: Predicted CDR loops (from tools like AbLang-2 or ImmuneBUilder) can be used as active residues for the antibody partner. Experimentally identified epitope residues serve as active restraints for the antigen. Multi-chain antibody structures (VH + VL) are collapsed into one docking body by the compatibility safeguard described above.
Cross-linking mass spectrometry integration: Cross-link evidence can be represented through the active/passive residue interface fields. Direct arbitrary unambiguous-restraint file upload is not currently available in this form.
The method assumes proteins maintain their unbound conformations. Semi-flexible refinement allows local side-chain and backbone adjustments at the interface, but large conformational changes upon binding are not captured. If the complex involves intrinsic disorder, coiled-coil rearrangements, or domain-level conformational shifts, accuracy will be limited regardless of restraint quality.
Computational time scales with sampling count. A run with 50 rigid-body models typically completes in 15-30 minutes; 200 models with full refinement can take 1-2 hours.
HADDOCK3 requires protein atoms in both uploaded partners. Non-standard ligands, cofactors, and unsupported glycans require both a CNS topology file and a CNS parameter file, which can be uploaded in the optional custom-ligand inputs. The maintained HADDOCK3 PDB preprocessor selects alternate locations, normalizes records, and removes CONECT records; custom connectivity must therefore be represented in the CNS files.
When active or passive residue restraints are used, PDB insertion codes must be normalized first. HADDOCK3 preprocessing can renumber insertion-coded and downstream residues, so ProteinIQ rejects this combination instead of risking restraints that target the wrong residues.
TIMEOUT_ERROR: Job exceeded runtime limits. Reduce rigid-body sampling, the pre-refinement selection count, or models retained per cluster.NO_COMPLEXES_GENERATED: HADDOCK3 finished but produced no models. Validate input PDB quality with PDB Fixer and consider adding interface restraints.INVALID_RESTRAINT_SELECTION: Chain or residue IDs in restraint fields do not match the preprocessed structures. Leave chain fields blank for automatic selection, or provide valid IDs from the input PDB.MODULE_OUTPUT_GENERATION_ERROR: A specific workflow stage (usually rigidbody or flexref) failed to produce any models within the tolerance. For multi-chain inputs, check whether chains were processed correctly. For single-chain inputs with restraints, verify residue numbers match the PDB exactly.HADDOCK_EXECUTION_ERROR: Generic CNS runtime failure. Retry with safer defaults; contact support if it repeats.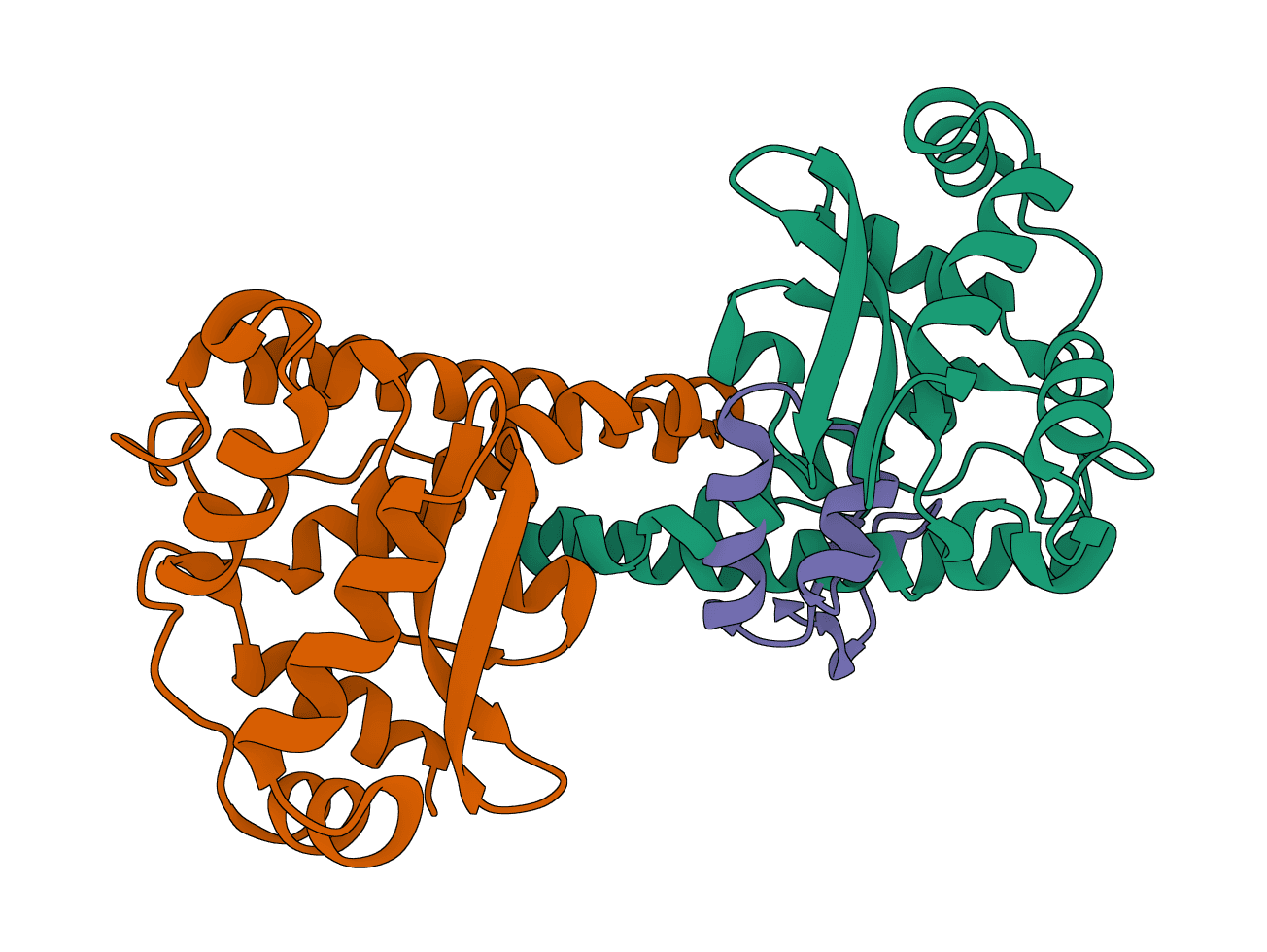
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
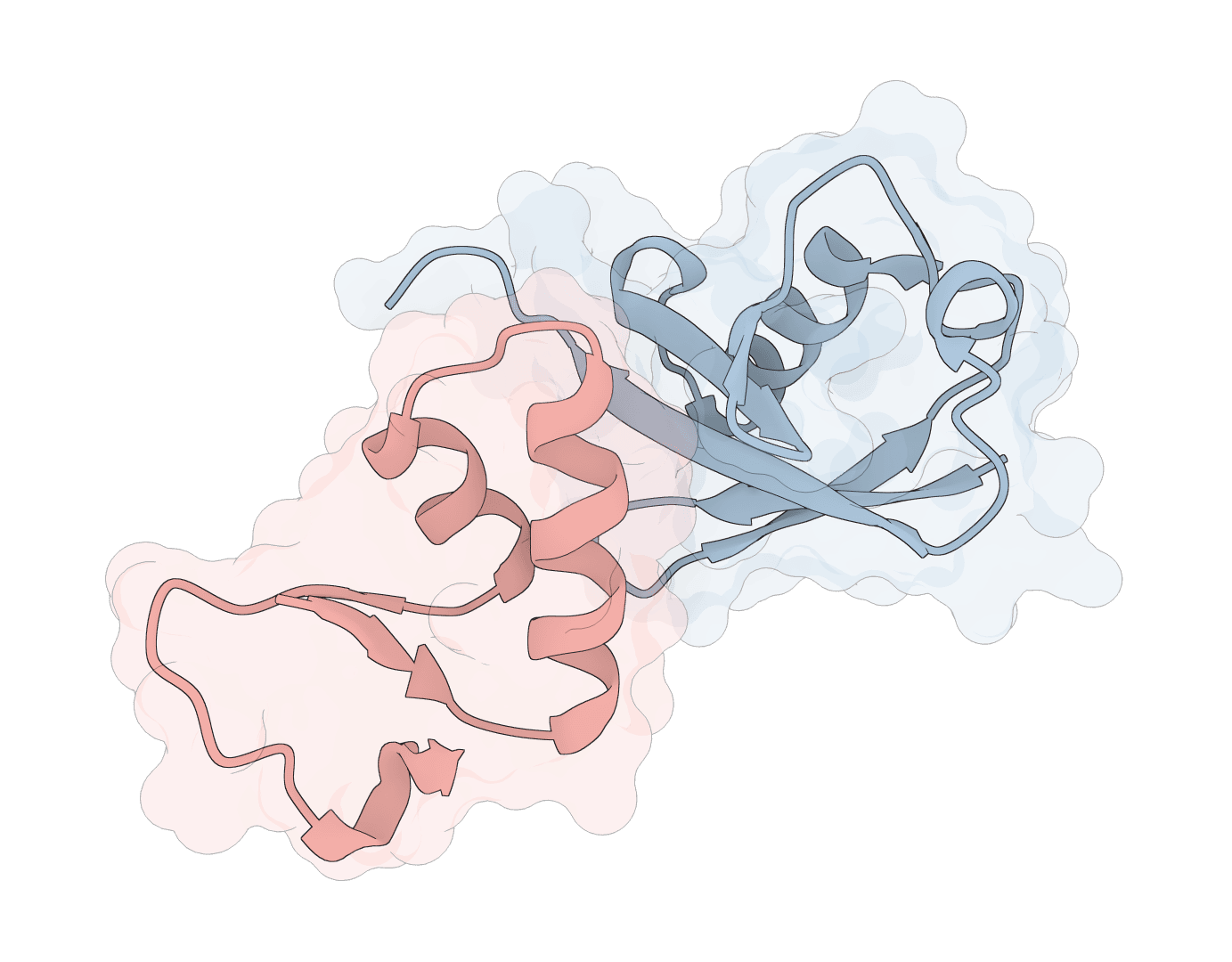
AF2Dock adapts AlphaFold2-style co-folding for structure-based protein-protein docking. It docks receptor and ligand protein structures with flow-matching refinement and ranks sampled complexes by iPTM.
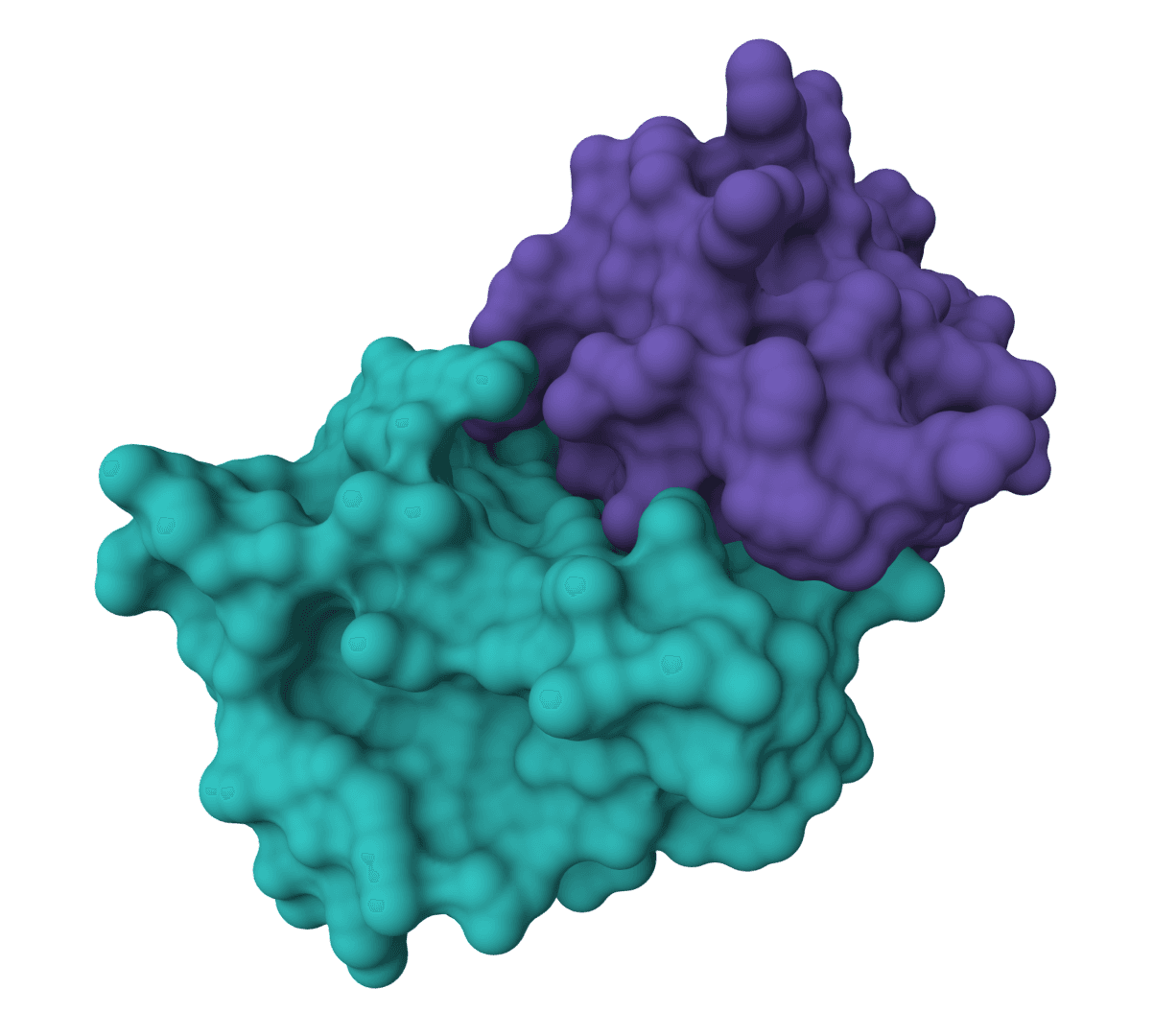
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.

DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
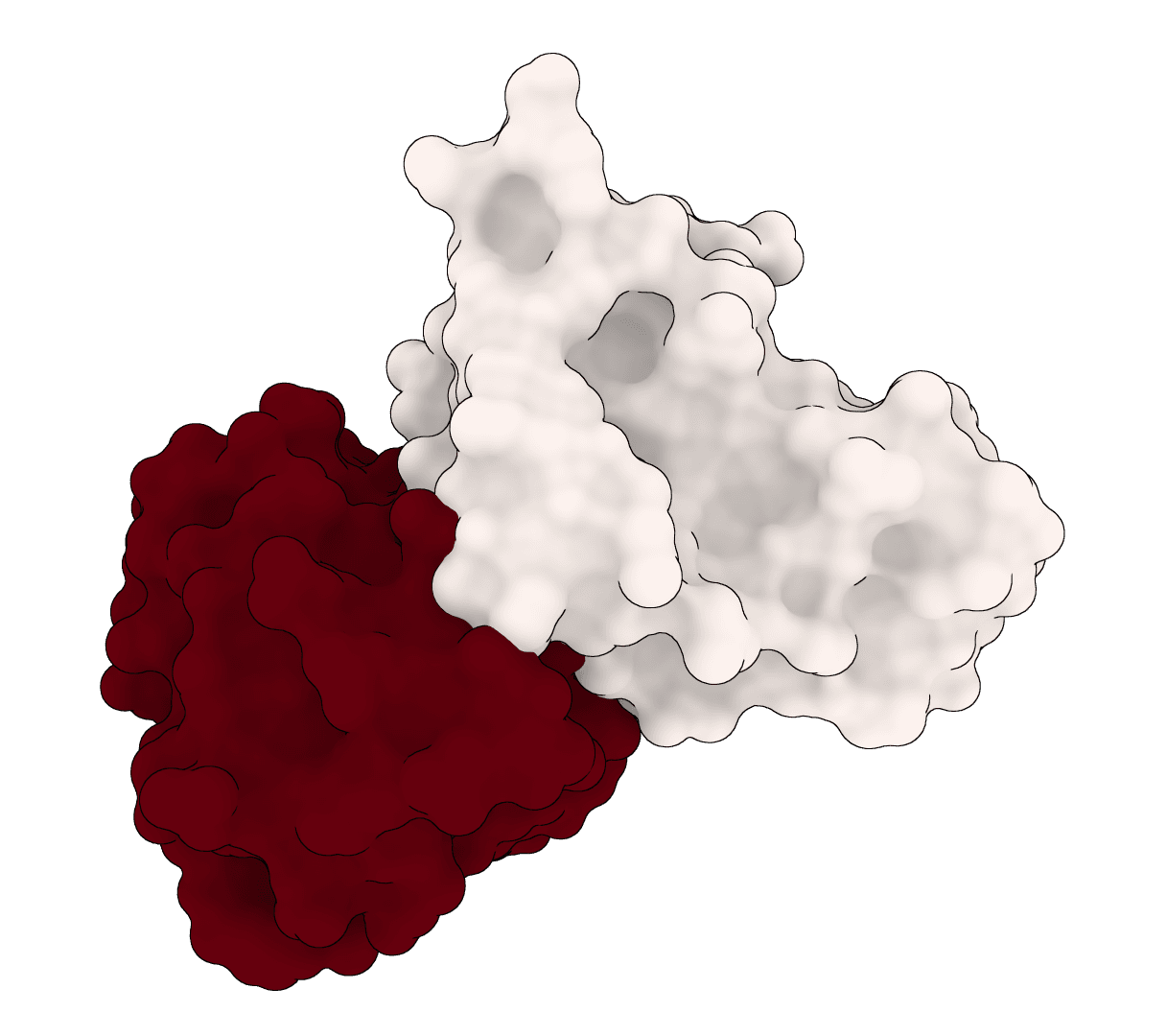
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
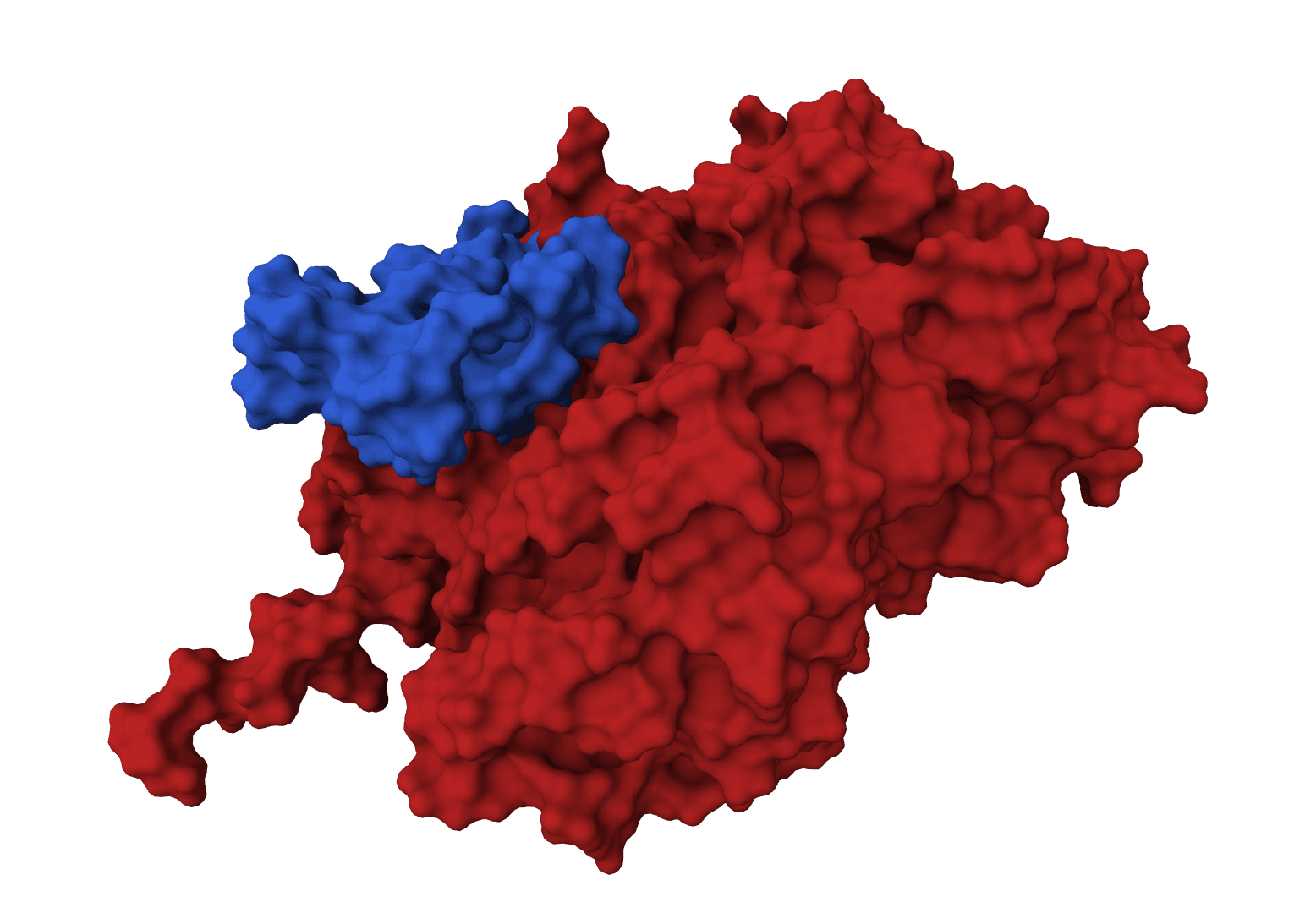
GeoDock predicts flexible protein-protein docking complexes from two separate protein structures using a multi-track iterative transformer and the DIPS 0.3 checkpoint from the Gray Lab release.
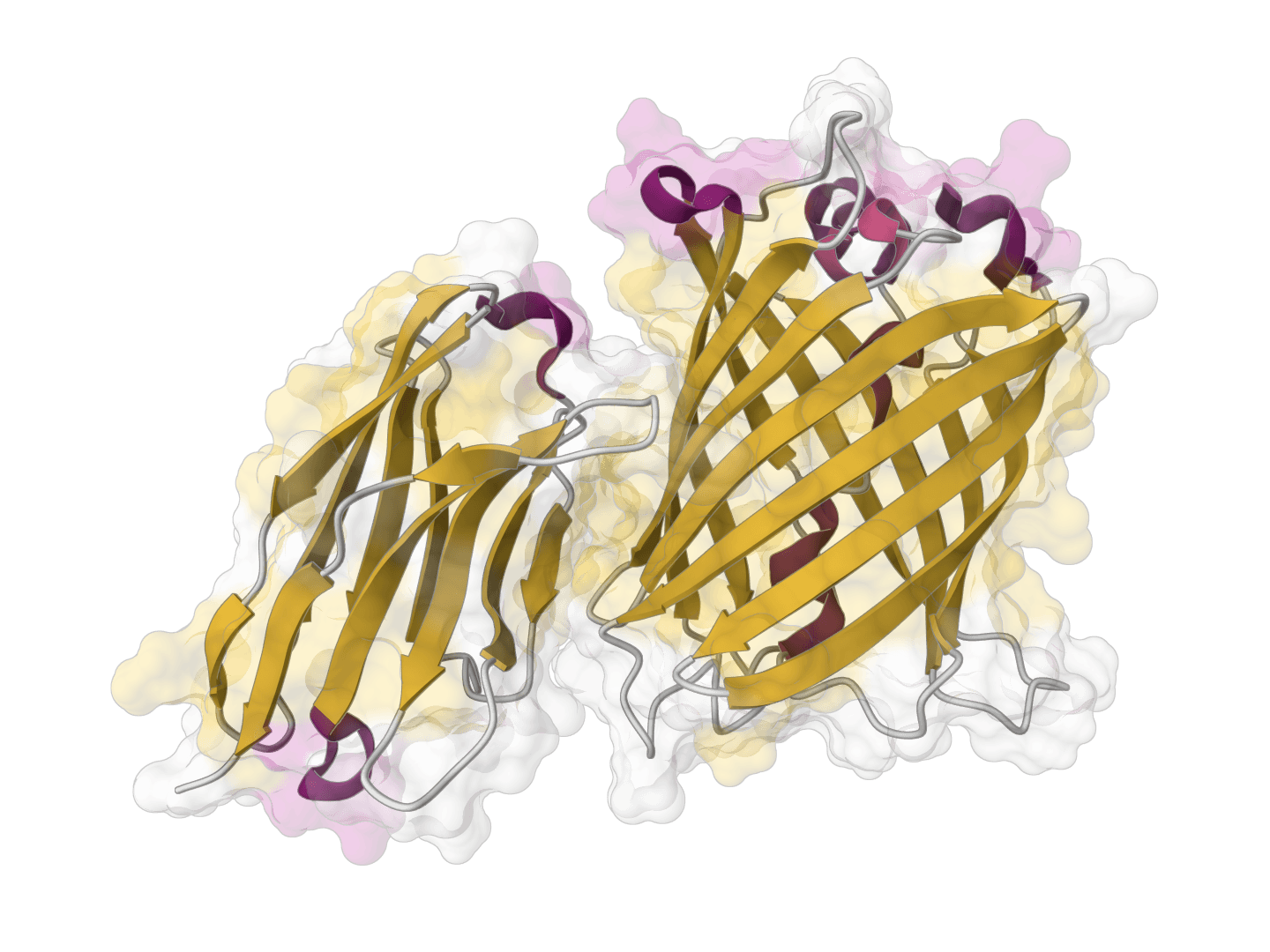
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
HADDOCK3 (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for predicting the structure of biomolecular complexes. Unlike docking methods that rely solely on shape complementarity, HADDOCK3 incorporates experimental or bioinformatic data as restraints to guide the search. This makes it particularly effective when partial interface information is available from mutagenesis, NMR chemical shift perturbations, cross-linking mass spectrometry, or epitope mapping.
HADDOCK3 is developed by the Bonvin Lab at Utrecht University and represents a complete redesign of the original HADDOCK platform with a fully modular workflow.
Upload two protein structures (PDB or mmCIF format, or enter a PDB ID) as the receptor and ligand, optionally provide interface residue numbers for each partner, and submit the job. ProteinIQ returns ranked docked complexes, the complete per-model and per-cluster CAPRI tables, source energy terms, clustering files, the effective configuration, the run log, and a bounded native analysis archive.
| Input | Format | Notes |
|---|---|---|
Receptor | PDB, ENT, CIF, mmCIF, or PDB ID | Protein atoms required. mmCIF inputs are converted to PDB automatically. |
Ligand | PDB, ENT, CIF, mmCIF, or PDB ID | The second docking partner, typically the smaller protein. |
Active residues | Comma-separated numbers or ranges (e.g., 38,40,45 or 38-45) | Residues directly involved at the interface. |
Passive residues | Same format | Surface residues near active residues that may contribute to binding. |
Chain ID | Single letter | Leave blank to use the first detected chain after preprocessing. |
| Setting | Default | Description |
|---|---|---|
Number of models | 50 | Rigid-body sampling count. Start with 50; use 100-200 with reliable restraints. |
Top models to return | 10 | Final complexes returned after clustering and refinement. |
Ab initio mode | Center-of-mass | Fallback guidance when no restraints are provided. cmrest applies center-of-mass restraints; ranair uses random AIRs; none docks freely. |
CPU cores | 8 | Parallelization used by HADDOCK3 inside the 8-core runtime. |
Rigid body tolerance | 5 | Percentage of failed rigidbody models accepted before the run aborts (default). |
Flexref tolerance | 5 | Same as above for the semi-flexible refinement stage. |
EM refinement tolerance | 5 | Same for energy minimization. |
Select top models before refinement | Automatic | Uses the HADDOCK3 default of 200, capped at the rigid-body sampling count. |
Minimum cluster population | 1 | Clusters with fewer members than this are discarded. Set low for small sampling counts. |
Models retained per cluster | 10 | Models retained from each selected cluster for CAPRI evaluation. This is separate from the total number returned. |
Each returned complex includes its HADDOCK score, native energy terms, cluster identity, and CAPRI fields. Because the current form does not accept an experimental reference complex, RMSD, Fnat, and DockQ values are relative to HADDOCK3's lowest-scoring run model, not an experimentally determined native structure.
| Column | Description |
|---|---|
rank | Global rank by HADDOCK score (lower is better). |
score | Weighted HADDOCK score in arbitrary units. |
vdw | Intermolecular van der Waals energy (kcal/mol). |
elec | Intermolecular electrostatic energy (kcal/mol). |
desolv | Empirical desolvation energy (kcal/mol). |
air | Ambiguous interaction restraint violation energy (kcal/mol). |
bsa | Buried surface area at the interface (Ų). |
cluster | Cluster identifier. Models in the same cluster adopt similar interface conformations. |
dockq, irmsd, fnat, lrmsd, ilrmsd | CAPRI comparison metrics relative to the lowest-scoring run model. |
total, angles, bonds, cdih, coup, dani, dihe, improper, rdcs, rg, vean, xpcs | Additional native energy and structural terms returned by CAPRI evaluation. |
The Files tab also includes the untouched capri_ss.tsv, capri_clt.tsv, FCC clustering tables, effective workflow configuration, run log, bounded analysis-report archive, and an artifact manifest. Coordinate and native-artifact payloads use aggregate limits; files omitted from an unusually large result are listed explicitly instead of being allowed to overload completion processing.
The core concept is the Ambiguous Interaction Restraint. An AIR encodes experimental uncertainty without requiring exact atomic contacts: it defines a distance restraint between any atom of an active residue on one protein and any atom on the active or passive residues of the partner. This flexibility accommodates the fact that most experimental techniques identify interface-proximal residues, not specific atomic contacts.
Active residues incur an energy penalty if they remain solvent-exposed in the docked complex. Passive residues are allowed but not required to participate. Random removal of a fraction of restraints during each docking trial ensures the search samples a range of conformations even when some restraints are incorrect.
The ProteinIQ implementation runs a five-stage pipeline:
Stage 1 - Topology generation (topoaa): Generates CNS-format all-atom topology for both proteins. Histidine protonation states are assigned automatically.
Stage 2 - Rigid body docking (rigidbody): Proteins are randomized in orientation and subjected to rigid-body energy minimization guided by AIRs or ab-initio restraints. The Number of models setting controls how many structures are generated here.
Stage 3 - Semi-flexible refinement (flexref): Top-scoring rigid-body models undergo simulated annealing in torsion angle space. Side chains and backbone atoms near the interface are free to move, optimizing local contacts without perturbing the global fold.
Stage 4 - Energy minimization refinement (emref): Selected models are refined by Cartesian energy minimization with electrostatics, van der Waals forces, and desolvation terms. This produces the final scored coordinates.
Stage 5 - Clustering and evaluation (clustfcc, seletopclusts, caprieval): Models are grouped by fraction of common contacts (FCC). Top models per cluster are selected, then scored by CAPRI evaluation, which computes the per-model energy breakdown and global ranking used in the results table.
The final HADDOCK score is a weighted sum of energy terms evaluated at the emref stage:
The heavy weight on van der Waals and desolvation reflects their central role in protein-protein recognition. Electrostatics is downweighted because its long-range character and sensitivity to dielectric treatment make it less reliable as a discriminator. Individual term values are returned alongside the composite score so that models can be compared by specific interaction type.
Typical scores for crystallographically validated complexes fall between -100 and -200. Absolute values scale with interface size, so ranking within a run is more informative than comparing scores across different protein pairs.
Both receptor and ligand accept multi-chain PDB files. A pinned-version compatibility reproducer showed that HADDOCK3's rigidbody CNS script can index more chain segments than docking partners, causing an undefined mol_fix_origin_N variable.
ProteinIQ retains the narrow compatibility workaround by collapsing each upload into a single docking body before the maintained HADDOCK3 preprocessor runs. Chain A is used for the receptor body and chain B for the ligand body. If interface residues are specified per chain, residue numbers are remapped onto the collapsed structure before source restraint generation. Multi-model PDB ensemble boundaries are preserved. When no restraints are provided for a multi-chain input, center-of-mass restraints (cmrest) are enabled automatically to maintain intermolecular guidance during rigid-body docking.
For single-chain inputs, overlapping chain IDs between receptor and ligand are remapped automatically (for example, if both use chain A, the ligand is reassigned to chain C before docking).
The vdw term is usually the most reliable discriminator between good and poor models. A very positive value (above +50) suggests steric clashes that were not resolved during refinement. The desolv term captures burial of hydrophobic surface; large negative values indicate a well-packed hydrophobic core. An air value significantly above zero means active residues are not forming the expected contacts, which can indicate incorrect restraints or a poorly sampled binding mode.
The buried surface area (bsa) is not part of the score formula but correlates with interface size. Values below 500 Ų typically indicate poor packing; values above 1500 Ų suggest large, well-formed interfaces.
Clusters with high population and low average score represent binding modes that are both energetically favorable and structurally consistent across multiple independent trials. When the top cluster contains most models, the docking has converged on a single binding mode. Fragmented clusters with similar scores suggest an underdefined interface, where increasing sampling or adding restraints would help.
If restraints are reliable, the top-ranked model within the largest cluster is most likely to approximate the native binding mode. For completely unknown interfaces (ab initio mode), the top 2-3 clusters should be examined since multiple modes may be plausible.
Data-driven docking uses active and passive residues to focus the search on known interface regions. Success rates exceed 70% in benchmark studies when restraints are correctly assigned. Convergence is faster because the rigid-body sampling is not exploring the full translational and rotational space.
Ab initio docking runs when no restraints are provided. HADDOCK3 generates random AIRs automatically (ranair), or restricts sampling using center-of-mass restraints (cmrest). Success rates drop to 30-40%, but the mode is useful when no interface information is available or when testing whether experimental observations are consistent with a proposed binding site.
Center-of-mass restraints (cmrest, the default) represent the best trade-off for ab initio runs: they ensure the proteins come into proximity without biasing which surface forms the interface.
HADDOCK3 is the right choice when some experimental interface information is available. The data-driven mode consistently outperforms purely computational approaches for these cases. It handles protein-protein complexes of arbitrary size, including antibody-antigen, enzyme-substrate, and homo-oligomeric systems.
LightDock is faster for exploratory runs with no interface data, using swarm intelligence to sample a wide range of docking poses. It does not support AIRs natively, so it loses the advantage HADDOCK3 has when restraints are available.
ColabDock and EquiDock are deep learning approaches that require no restraints and run faster, but cannot incorporate experimental data the way HADDOCK3 can.
For protein-ligand docking (small molecule into a protein binding site), use AutoDock Vina, GNINA, or DiffDock instead. HADDOCK3 is designed for macromolecular interfaces.
NMR-based complex modeling: Chemical shift perturbations identify residues affected by binding. Map perturbed residues to active, surface-adjacent residues to passive, and submit. HADDOCK3's accuracy in this regime is well established across hundreds of benchmark complexes.
Mutagenesis-guided docking: Mutations that disrupt binding localize the interface. Alanine scan results map directly to active residues. Surrounding exposed surface residues serve as passive restraints.
Antibody-antigen docking: Predicted CDR loops (from tools like AbLang-2 or ImmuneBUilder) can be used as active residues for the antibody partner. Experimentally identified epitope residues serve as active restraints for the antigen. Multi-chain antibody structures (VH + VL) are collapsed into one docking body by the compatibility safeguard described above.
Cross-linking mass spectrometry integration: Cross-link evidence can be represented through the active/passive residue interface fields. Direct arbitrary unambiguous-restraint file upload is not currently available in this form.
The method assumes proteins maintain their unbound conformations. Semi-flexible refinement allows local side-chain and backbone adjustments at the interface, but large conformational changes upon binding are not captured. If the complex involves intrinsic disorder, coiled-coil rearrangements, or domain-level conformational shifts, accuracy will be limited regardless of restraint quality.
Computational time scales with sampling count. A run with 50 rigid-body models typically completes in 15-30 minutes; 200 models with full refinement can take 1-2 hours.
HADDOCK3 requires protein atoms in both uploaded partners. Non-standard ligands, cofactors, and unsupported glycans require both a CNS topology file and a CNS parameter file, which can be uploaded in the optional custom-ligand inputs. The maintained HADDOCK3 PDB preprocessor selects alternate locations, normalizes records, and removes CONECT records; custom connectivity must therefore be represented in the CNS files.
When active or passive residue restraints are used, PDB insertion codes must be normalized first. HADDOCK3 preprocessing can renumber insertion-coded and downstream residues, so ProteinIQ rejects this combination instead of risking restraints that target the wrong residues.
TIMEOUT_ERROR: Job exceeded runtime limits. Reduce rigid-body sampling, the pre-refinement selection count, or models retained per cluster.NO_COMPLEXES_GENERATED: HADDOCK3 finished but produced no models. Validate input PDB quality with PDB Fixer and consider adding interface restraints.INVALID_RESTRAINT_SELECTION: Chain or residue IDs in restraint fields do not match the preprocessed structures. Leave chain fields blank for automatic selection, or provide valid IDs from the input PDB.MODULE_OUTPUT_GENERATION_ERROR: A specific workflow stage (usually rigidbody or flexref) failed to produce any models within the tolerance. For multi-chain inputs, check whether chains were processed correctly. For single-chain inputs with restraints, verify residue numbers match the PDB exactly.HADDOCK_EXECUTION_ERROR: Generic CNS runtime failure. Retry with safer defaults; contact support if it repeats.LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
AF2Dock adapts AlphaFold2-style co-folding for structure-based protein-protein docking. It docks receptor and ligand protein structures with flow-matching refinement and ranks sampled complexes by iPTM.
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
GeoDock predicts flexible protein-protein docking complexes from two separate protein structures using a multi-track iterative transformer and the DIPS 0.3 checkpoint from the Gray Lab release.
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.