Predict mutation ΔΔG values and identify stabilizing substitutions for protein engineering. Learn more
Run
20 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job”.
Predict mutation ΔΔG values and identify stabilizing substitutions for protein engineering. Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
Predict mutation ΔΔG values and identify stabilizing substitutions for protein engineering. Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
ThermoMPNN is a graph neural network that predicts how single amino acid mutations affect protein thermostability. Developed by Henry Dieckhaus and colleagues at the Kuhlman Lab (University of North Carolina), ThermoMPNN predicts ΔΔG (change in free energy of folding) for point mutations, enabling rapid identification of stabilizing or destabilizing mutations.
The model employs transfer learning from ProteinMPNN, a pretrained sequence recovery model. Rather than training a stability predictor from scratch, ThermoMPNN extracts learned structural embeddings from ProteinMPNN and fine-tunes a lightweight prediction module on stability data. This approach achieves state-of-the-art performance while remaining computationally efficient.
ThermoMPNN was published in Proceedings of the National Academy of Sciences in 2024 and trained on the Megascale dataset containing over 270,000 stability measurements.
The ThermoMPNN family has expanded since its initial release:
ProteinIQ runs ThermoMPNN's documented analysis/custom_inference.py entry point at a pinned source commit. Upload a protein structure, specify one chain to analyze, and receive ΔΔG predictions for all possible single mutations at each position (site-saturation mutagenesis).
| Input | Description |
|---|---|
Protein Structure | The protein to analyze. Upload a PDB file or enter a PDB ID (e.g., 1HSG) to fetch from RCSB. |
| Setting | Description |
|---|---|
Chain to analyze | Which chain to run predictions on. The custom_inference.py default is chain A; leaving this blank auto-selects the first chain in the structure. |
The primary output is the native custom_inference.py CSV for every possible mutation at each sequence position. The submitted structure name is preserved in the native dataset label and filename. A JSON provenance file records the exact source commit and model checksum used for the run.
| Column | Description |
|---|---|
Model | published model label, ThermoMPNN. |
Dataset | Input PDB dataset label used by native inference. |
ddG_pred | Predicted change in folding free energy, displayed in kcal/mol. |
position | Zero-based sequence position used by native ThermoMPNN. |
wildtype | Original amino acid at this position (single-letter code). |
mutation | Substituted amino acid (single-letter code). |
pdb | PDB label used in the native output. |
chain | Chain identifier. |
The ΔΔG value represents the predicted change in thermodynamic stability upon mutation:
The model's dynamic range is approximately −5 to +5 kcal/mol based on its training data. Predictions outside this range should be interpreted with caution.
The output includes self-mutations (e.g., A→A) with ΔΔG values near zero. These serve as internal controls and confirm the model correctly predicts no stability change when the amino acid remains unchanged.
ThermoMPNN combines a frozen pretrained ProteinMPNN feature extractor with a lightweight stability prediction module. The model treats proteins as graphs where residues are nodes and spatial relationships between atoms define edges.
The architecture consists of three components:
ProteinMPNN feature extractor — A message-passing neural network with three encoder and three decoder layers. It processes structural information using Gaussian radial basis functions that encode distances to the 48 nearest neighboring residues. The encoder layers are frozen during training to preserve learned structural representations.
Light attention block — A self-attention mechanism with padded convolutions that reweights the extracted embeddings based on learned context. This allows the model to focus on residue features most relevant to stability prediction.
MLP prediction head — A multilayer perceptron with two hidden layers (sizes 64 and 32) that outputs ΔΔG predictions. The final value is computed by subtracting the predicted ΔG for the wild-type amino acid from the predicted ΔG of the mutant amino acid.
Traditional stability predictors require large amounts of experimental stability data for training. ThermoMPNN circumvents this limitation by leveraging ProteinMPNN's pretrained knowledge of protein structure-sequence relationships. The ProteinMPNN encoder has learned generalizable structural features from millions of protein sequences, which transfer effectively to stability prediction tasks.
The primary training dataset is the Megascale dataset from Tsuboyama et al., containing 272,712 stability measurements across 298 proteins (181 natural and 109 de novo designed). These measurements derive from proteolysis sensitivity experiments with a dynamic range of approximately 5 kcal/mol.
The model was additionally validated on the Fireprot dataset (3,438 mutations across 100 proteins), which contains traditional biophysical measurements with a wider dynamic range (−9 to +12 kcal/mol).
Benchmark performance on held-out test sets:
| Dataset | Pearson Correlation | RMSE (kcal/mol) |
|---|---|---|
| Megascale | 0.754 | 0.708 |
| Fireprot (homologue-free) | 0.650 | 1.51 |
| Ssym (direct) | 0.72 | — |
For identifying stabilizing mutations (ΔΔG < −0.5 kcal/mol), the positive predictive value is approximately 56% on Fireprot and 46% on Megascale.
Dynamic range constraint — Training on Megascale limits accurate predictions to approximately ±5 kcal/mol. Larger stability changes may show degraded performance.
Epistatic effects — Single-mutation predictions assume additive effects. A 2025 study in Protein Science demonstrated that stability models, including ThermoMPNN, struggle to capture epistatic interactions of double point mutations. For multiple mutations, consider using ThermoMPNN-D or validating experimentally.
Surface cysteine artifacts — The Megascale assay methodology artificially favors surface cysteines through intermolecular disulfide formation. Cysteine predictions at surface positions should be interpreted cautiously.
Hydrophobicity bias — The model exhibits a slight bias toward hydrophobic mutations, which could promote aggregation if used for comprehensive protein redesign rather than targeted single-site optimization.
Structure quality dependency — Performance on low-confidence structures (pLDDT < 0.75) or NMR structures may be reduced compared to high-resolution crystal structures.
Single mutations only — ThermoMPNN predicts effects of individual point mutations. For double mutations with epistatic effects, ThermoMPNN-D is available (separate tool).
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.

Predict protein stability using validated BioPython methods: Instability Index, Aliphatic Index, GRAVY, flexibility analysis, and charge distribution
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
Predict protein solubility from amino acid sequence using the University of Manchester Protein-Sol method.
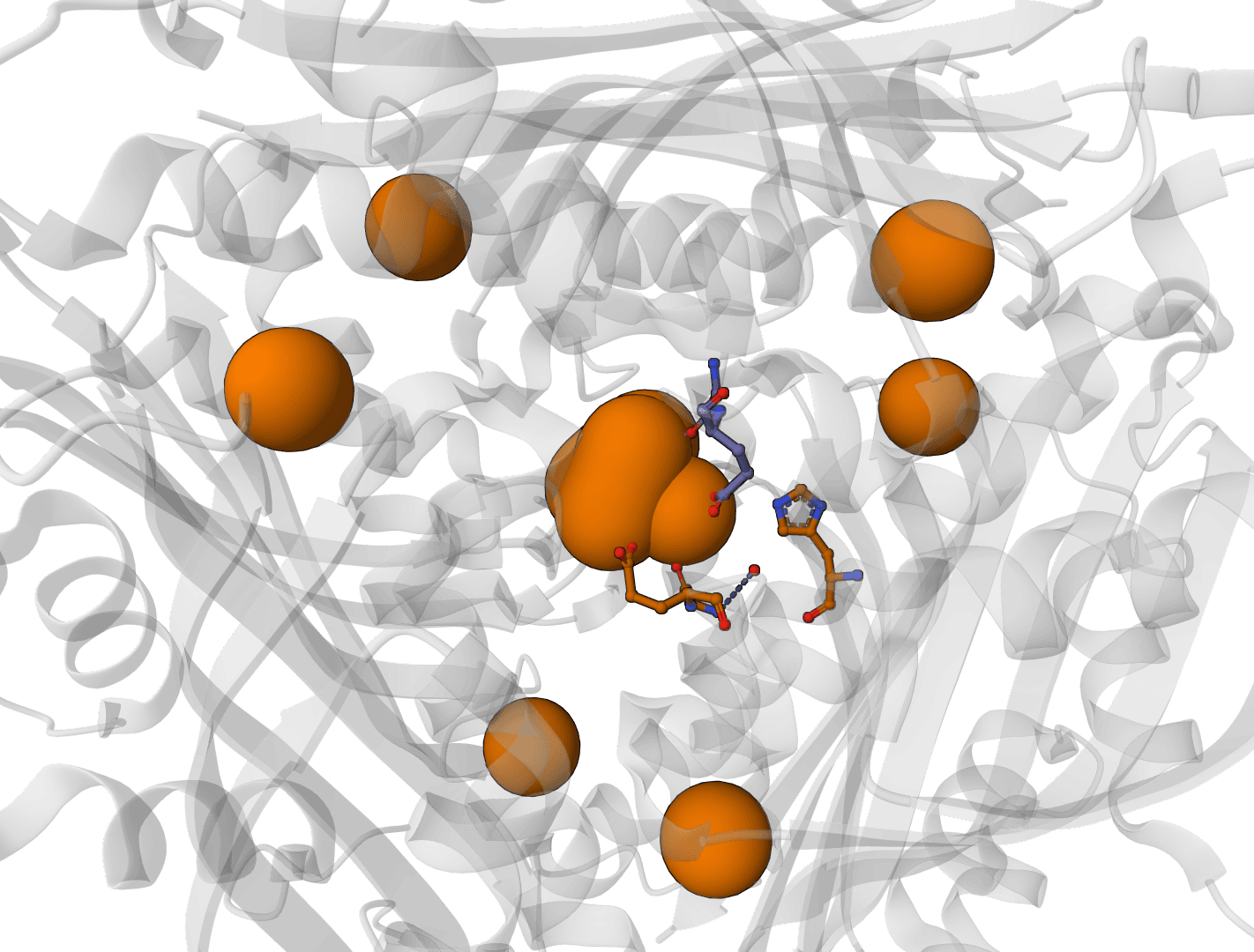
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
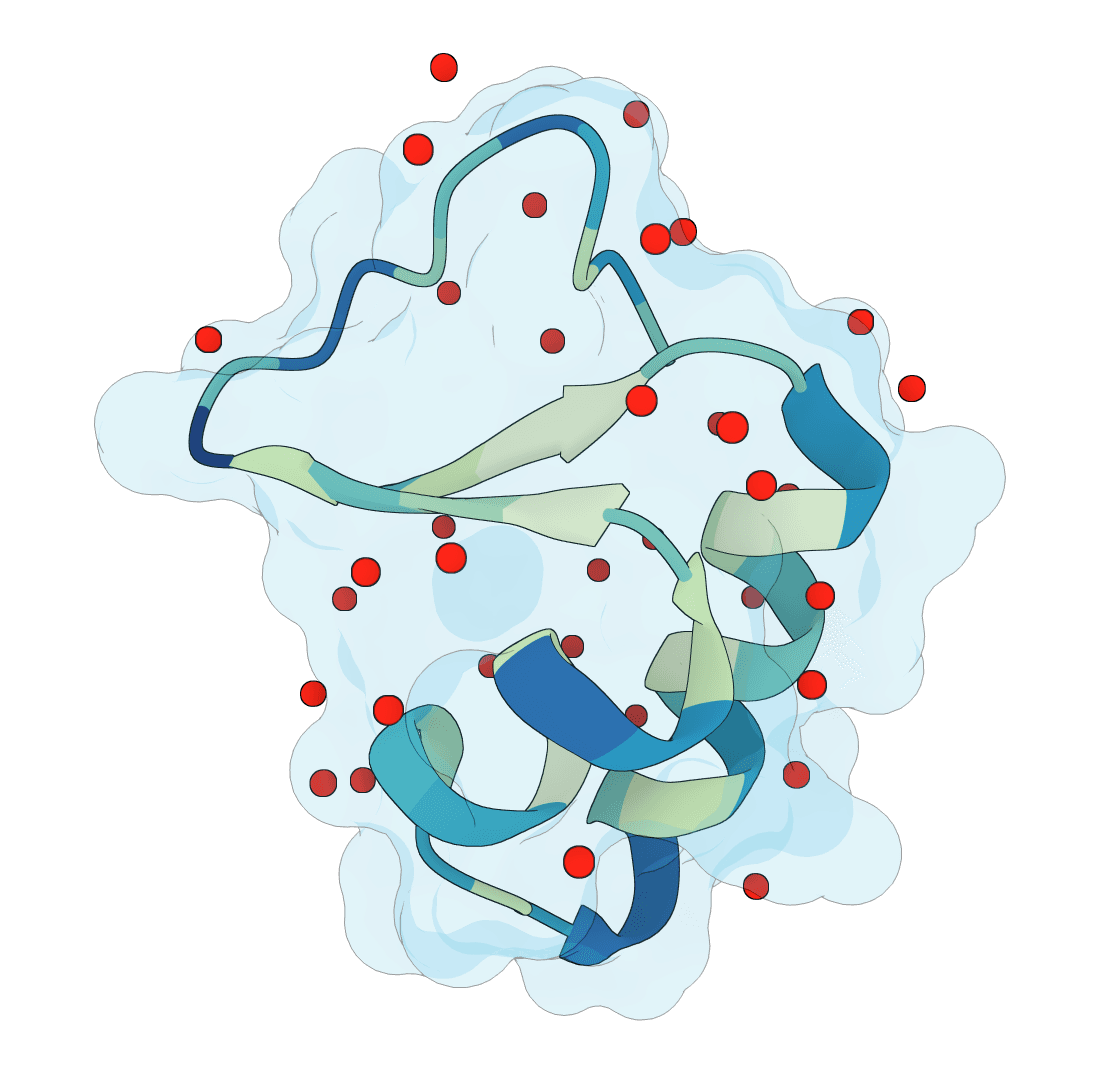
Predict protein hydration sites from a structure using a diffusion model with ESM features and a confidence-filtering head.
Compute 200+ RDKit molecular descriptors, drug-likeness rule violations, and structural fingerprints for QSAR, virtual screening, and ML workflows
ThermoMPNN is a graph neural network that predicts how single amino acid mutations affect protein thermostability. Developed by Henry Dieckhaus and colleagues at the Kuhlman Lab (University of North Carolina), ThermoMPNN predicts ΔΔG (change in free energy of folding) for point mutations, enabling rapid identification of stabilizing or destabilizing mutations.
The model employs transfer learning from ProteinMPNN, a pretrained sequence recovery model. Rather than training a stability predictor from scratch, ThermoMPNN extracts learned structural embeddings from ProteinMPNN and fine-tunes a lightweight prediction module on stability data. This approach achieves state-of-the-art performance while remaining computationally efficient.
ThermoMPNN was published in Proceedings of the National Academy of Sciences in 2024 and trained on the Megascale dataset containing over 270,000 stability measurements.
The ThermoMPNN family has expanded since its initial release:
ProteinIQ runs ThermoMPNN's documented analysis/custom_inference.py entry point at a pinned source commit. Upload a protein structure, specify one chain to analyze, and receive ΔΔG predictions for all possible single mutations at each position (site-saturation mutagenesis).
| Input | Description |
|---|---|
Protein Structure | The protein to analyze. Upload a PDB file or enter a PDB ID (e.g., 1HSG) to fetch from RCSB. |
| Setting | Description |
|---|---|
Chain to analyze | Which chain to run predictions on. The custom_inference.py default is chain A; leaving this blank auto-selects the first chain in the structure. |
The primary output is the native custom_inference.py CSV for every possible mutation at each sequence position. The submitted structure name is preserved in the native dataset label and filename. A JSON provenance file records the exact source commit and model checksum used for the run.
| Column | Description |
|---|---|
Model | published model label, ThermoMPNN. |
Dataset | Input PDB dataset label used by native inference. |
ddG_pred | Predicted change in folding free energy, displayed in kcal/mol. |
position | Zero-based sequence position used by native ThermoMPNN. |
wildtype | Original amino acid at this position (single-letter code). |
mutation | Substituted amino acid (single-letter code). |
pdb | PDB label used in the native output. |
chain | Chain identifier. |
The ΔΔG value represents the predicted change in thermodynamic stability upon mutation:
The model's dynamic range is approximately −5 to +5 kcal/mol based on its training data. Predictions outside this range should be interpreted with caution.
The output includes self-mutations (e.g., A→A) with ΔΔG values near zero. These serve as internal controls and confirm the model correctly predicts no stability change when the amino acid remains unchanged.
ThermoMPNN combines a frozen pretrained ProteinMPNN feature extractor with a lightweight stability prediction module. The model treats proteins as graphs where residues are nodes and spatial relationships between atoms define edges.
The architecture consists of three components:
ProteinMPNN feature extractor — A message-passing neural network with three encoder and three decoder layers. It processes structural information using Gaussian radial basis functions that encode distances to the 48 nearest neighboring residues. The encoder layers are frozen during training to preserve learned structural representations.
Light attention block — A self-attention mechanism with padded convolutions that reweights the extracted embeddings based on learned context. This allows the model to focus on residue features most relevant to stability prediction.
MLP prediction head — A multilayer perceptron with two hidden layers (sizes 64 and 32) that outputs ΔΔG predictions. The final value is computed by subtracting the predicted ΔG for the wild-type amino acid from the predicted ΔG of the mutant amino acid.
Traditional stability predictors require large amounts of experimental stability data for training. ThermoMPNN circumvents this limitation by leveraging ProteinMPNN's pretrained knowledge of protein structure-sequence relationships. The ProteinMPNN encoder has learned generalizable structural features from millions of protein sequences, which transfer effectively to stability prediction tasks.
The primary training dataset is the Megascale dataset from Tsuboyama et al., containing 272,712 stability measurements across 298 proteins (181 natural and 109 de novo designed). These measurements derive from proteolysis sensitivity experiments with a dynamic range of approximately 5 kcal/mol.
The model was additionally validated on the Fireprot dataset (3,438 mutations across 100 proteins), which contains traditional biophysical measurements with a wider dynamic range (−9 to +12 kcal/mol).
Benchmark performance on held-out test sets:
| Dataset | Pearson Correlation | RMSE (kcal/mol) |
|---|---|---|
| Megascale | 0.754 | 0.708 |
| Fireprot (homologue-free) | 0.650 | 1.51 |
| Ssym (direct) | 0.72 | — |
For identifying stabilizing mutations (ΔΔG < −0.5 kcal/mol), the positive predictive value is approximately 56% on Fireprot and 46% on Megascale.
Dynamic range constraint — Training on Megascale limits accurate predictions to approximately ±5 kcal/mol. Larger stability changes may show degraded performance.
Epistatic effects — Single-mutation predictions assume additive effects. A 2025 study in Protein Science demonstrated that stability models, including ThermoMPNN, struggle to capture epistatic interactions of double point mutations. For multiple mutations, consider using ThermoMPNN-D or validating experimentally.
Surface cysteine artifacts — The Megascale assay methodology artificially favors surface cysteines through intermolecular disulfide formation. Cysteine predictions at surface positions should be interpreted cautiously.
Hydrophobicity bias — The model exhibits a slight bias toward hydrophobic mutations, which could promote aggregation if used for comprehensive protein redesign rather than targeted single-site optimization.
Structure quality dependency — Performance on low-confidence structures (pLDDT < 0.75) or NMR structures may be reduced compared to high-resolution crystal structures.
Single mutations only — ThermoMPNN predicts effects of individual point mutations. For double mutations with epistatic effects, ThermoMPNN-D is available (separate tool).
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.
Predict protein stability using validated BioPython methods: Instability Index, Aliphatic Index, GRAVY, flexibility analysis, and charge distribution
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
Predict protein solubility from amino acid sequence using the University of Manchester Protein-Sol method.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Predict protein hydration sites from a structure using a diffusion model with ESM features and a confidence-filtering head.
Compute 200+ RDKit molecular descriptors, drug-likeness rule violations, and structural fingerprints for QSAR, virtual screening, and ML workflows