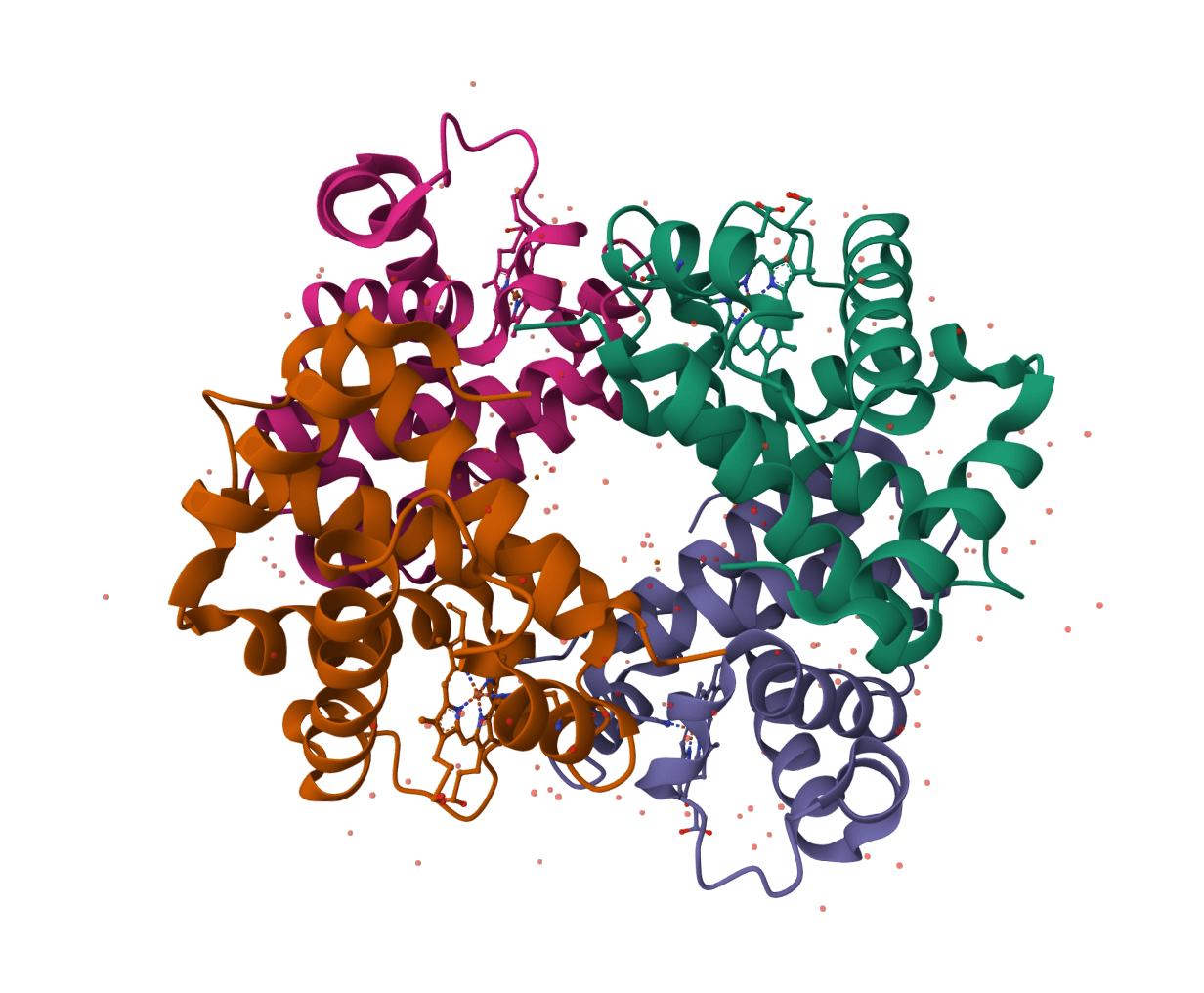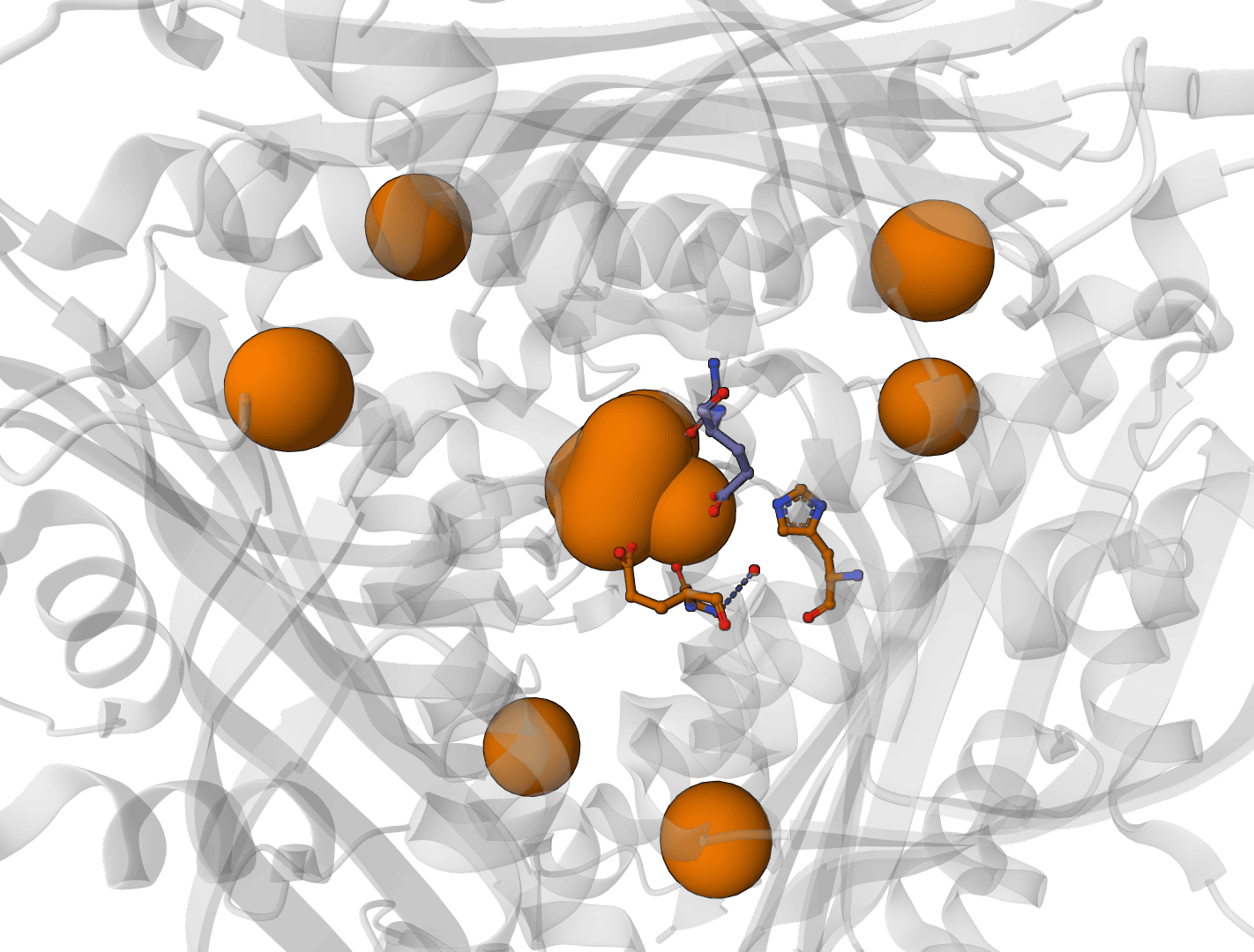What is PDBsum?
PDBsum turns a protein structure into a browsable structural summary. The report collects diagrams, interaction views, validation-oriented pages, logs, and supporting assets so a structure can be reviewed as a connected HTML report rather than as a raw coordinate file.
PDBsum1 is the standalone program behind this analysis. It generates the same type of analyses as the public PDBsum Generate service and gives direct access to the computed result files. That makes it useful for reviewing deposited PDB structures, checking modeled structures before downstream analysis, and packaging structure summaries for collaboration.
How to use PDBsum online
Run PDBsum online by uploading one protein structure in PDB or ENT format, including gzipped files, or by fetching a structure from RCSB with a PDB ID. ProteinIQ analyzes the structure as a single PDBsum1 job and returns a downloadable ZIP archive containing the generated report directory.
PDBsum expects correct PDB-format coordinate records. It can tolerate some common formatting problems, but files from docking, modeling, or structure-editing tools may still fail if atom records, chain identifiers, residue names, or header records are malformed.
For best reports, include useful TITLE and DBREF records when working with custom files. TITLE helps label the generated report. DBREF can help PDBsum connect the structure to UniProt and domain information when those records are available.
Settings
Results
PDBsum returns a report archive, not a spreadsheet. The output list contains one ZIP file with the complete generated result directory.
The archive can include:
- HTML report pages: Entry pages, index files, and linked report sections generated by PDBsum
- Diagrams and assets: Images and supporting files used by the HTML pages
- Logs: Run logs and diagnostic files generated during analysis
- Viewer scripts: RasMol scripts when
Include RasMol scripts is enabled
Extract the ZIP before browsing the report so relative links between pages, images, and assets resolve correctly.
Understanding the report
PDBsum is strongest as a structure-review report. It is designed for quick inspection of what is in a coordinate file and how the structural features relate to each other.
The most useful pages depend on the structure:
- Protein overview: A compact summary of chains, domains, secondary structure, ligands, and linked structural annotations
- Interaction views: Diagrams for contacts involving ligands, DNA, RNA, proteins, or other bound components when those entities are present
- Clefts: Surface cleft analysis, useful for pocket inspection and ligand-binding context
- Validation-oriented pages: Structure-quality summaries and geometry checks where PDBsum generates them
- Raw generated files: Computed data and logs for users who need to inspect the exact files behind the report
The report should be read as a descriptive summary of the submitted coordinates. It does not repair a structure, add missing atoms, assign protonation states, or rescore a model. If the input coordinate file is incomplete or poorly formatted, the report can be incomplete or fail.
PDBsum is the right starting point when the goal is a broad visual summary of one protein structure. It is especially useful after fetching a PDB entry, receiving a modeled structure from a collaborator, or generating a new structure prediction that needs a quick structural overview.
Use MolProbity when the main question is stereochemical validation and model quality. MolProbity focuses on clashes, rotamers, Ramachandran statistics, and geometry checks.
Use DSSP when the main output needed is secondary-structure assignment rather than a full report. Use PDBFixer before PDBsum when a structure needs missing atoms, residues, or formatting problems fixed.
For protein complex evaluation, PDBsum can describe interfaces and contacts, but specialized scoring tools such as DockQ or ipSAE are better suited for quantitative complex-quality assessment.
Practical limitations
PDBsum analyzes one structure per ProteinIQ job. The standalone PDBsum1 program also supports batch lists, but this ProteinIQ tool keeps each job tied to one uploaded or fetched structure so the result archive and job history remain unambiguous.
Only PDB and ENT-style inputs are supported. mmCIF files should be converted to PDB before use with a structure-format converter that preserves the coordinate records PDBsum expects.
Large multi-chain structures can take longer when cleft calculation is enabled. Leave Calculate clefts for large structures off unless the cleft pages are needed for the analysis.