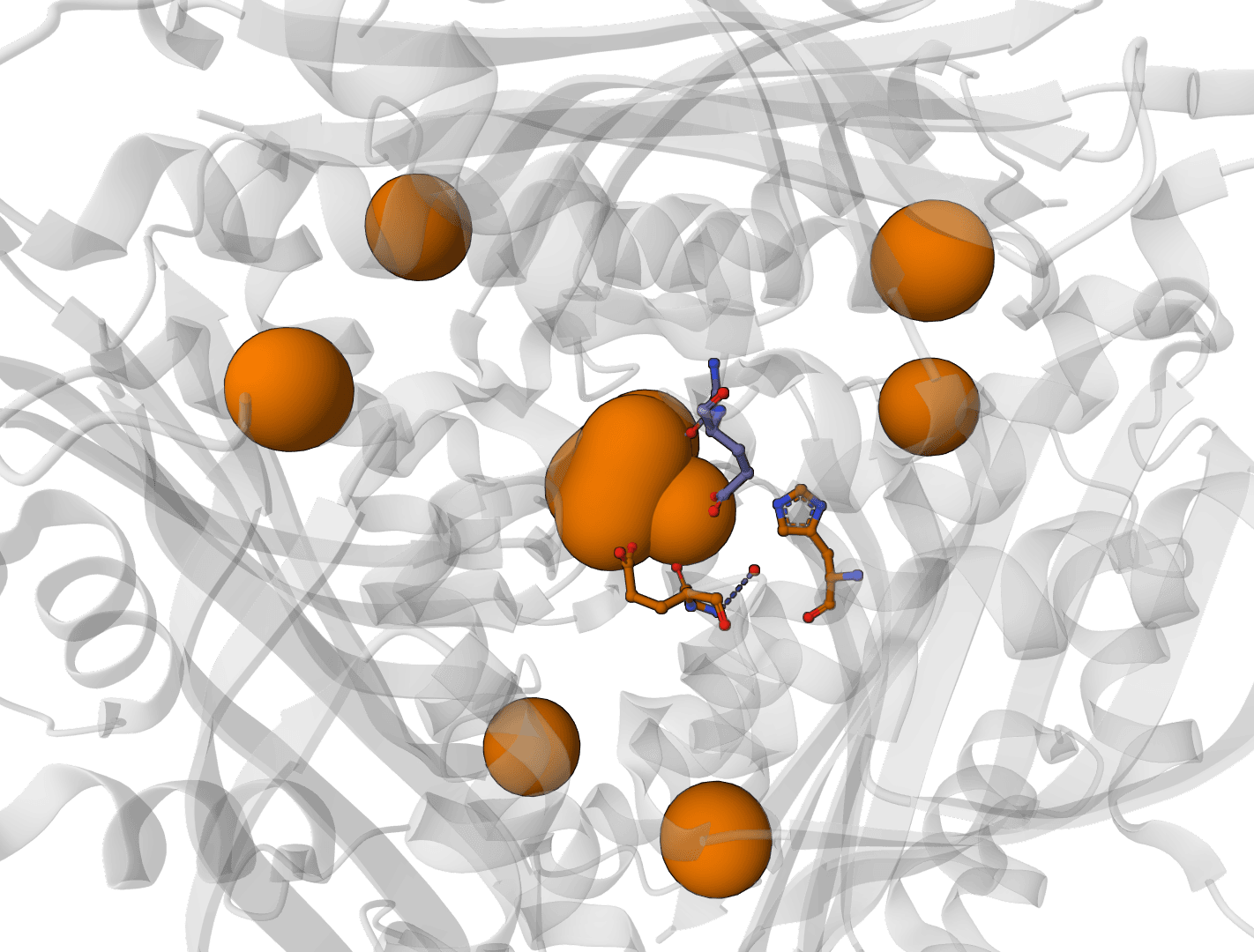
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Calculate pairwise RMSD matrices for PDB structure ensembles with pyRMSD, including the condensed matrix and source statistics files.
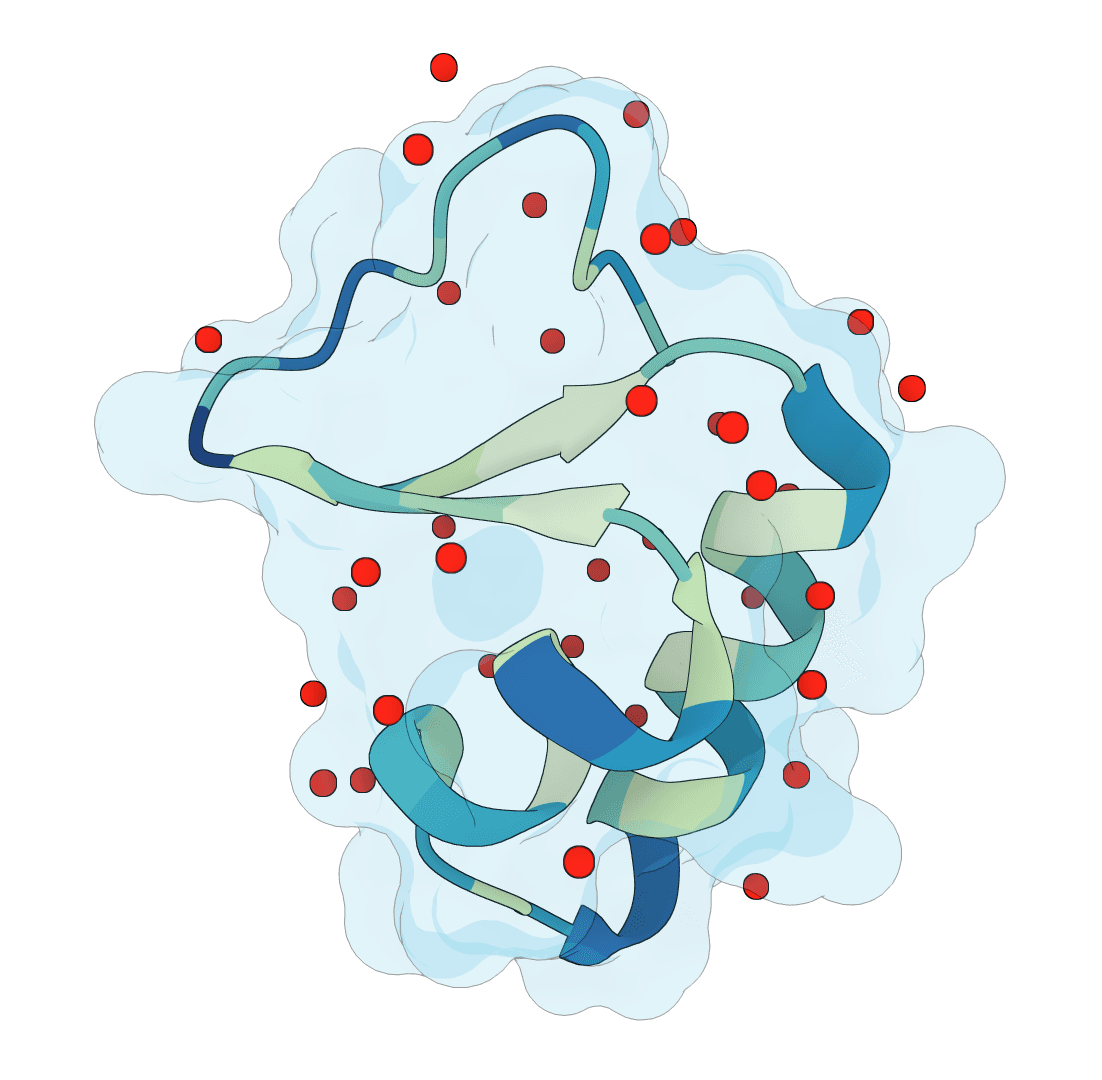
Predict protein hydration sites from a structure using a diffusion model with ESM features and a confidence-filtering head.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
Analyze noncovalent interactions in protein-ligand complex structures with PLIP, including hydrogen bonds, hydrophobic contacts, pi interactions, salt bridges, water bridges, halogen bonds, and metal complexes.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Calculate pairwise RMSD matrices for PDB structure ensembles with pyRMSD, including the condensed matrix and source statistics files.
Predict protein hydration sites from a structure using a diffusion model with ESM features and a confidence-filtering head.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
Analyze noncovalent interactions in protein-ligand complex structures with PLIP, including hydrogen bonds, hydrophobic contacts, pi interactions, salt bridges, water bridges, halogen bonds, and metal complexes.
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
TRPV1 half-maps — FSC-filtered local sharpening
β-galactosidase — full-map reference scaling
γ-secretase — masked and filtered full map
LocScale performs local density scaling for cryo-EM maps. It sharpens map regions according to local signal and reference information rather than applying one global B-factor across the whole volume.
ProteinIQ runs LocScale on uploaded MRC/MAP files and returns the sharpened map plus the processing log and generated map files. The first release focuses on the standard LocScale command for local sharpening with half-map or full-map inputs. Feature-enhanced maps, coordinate-model refinement, hybrid model completion, MPI runs, and PDF reports are tracked for future support.
| Input | Description |
|---|---|
| Half Map 1 | First unsharpened, unfiltered half-map in MRC or MAP format. |
| Half Map 2 | Second matching unsharpened, unfiltered half-map. |
| Cryo-EM Map | Full unsharpened map for datasets without half-maps. |
| Mask | Optional MRC/MAP mask. LocScale estimates an FDR mask when this is omitted. |
| Reference Map | Optional MRC/MAP model map used directly as the LocScale reference. |
Half-map input is recommended when available because LocScale can use the pair for FSC-based resolution estimation.
ProteinIQ accepts MRC/MAP uploads up to 500 MB per file for this service. This is a ProteinIQ upload limit, not a LocScale file-format limit.
The default settings match LocScale defaults for local sharpening: one CPU process, FDR threshold 0.01, averaging filter size 3, EMmerNet batch size 8, cube size 32, and no input filtering. Optional resolution, window-size, FDR-window, FDR pre-filter, mask-threshold, and reference-map-resolution fields are passed to the corresponding LocScale command-line options only when set.
LocScale returns a primary sharpened MRC map. ProteinIQ also returns the LocScale log and generated processing files such as FDR masks or local-scaling diagnostic maps when LocScale writes them.
Open the returned MRC files in ChimeraX, Coot, or another cryo-EM map viewer for inspection.
This example demonstrates the recommended paired-half-map workflow on a compact region of the Rattus norvegicus TRPV1 channel dataset EMD-5778. It uses matching unfiltered half-map subvolumes, a data-derived mask, and the corresponding deposited reconstruction as a direct reference map.
CPU processes = 4, Resolution cutoff (A) = 3.4, Window size (pixels) = 24, and Apply FSC filter = enabled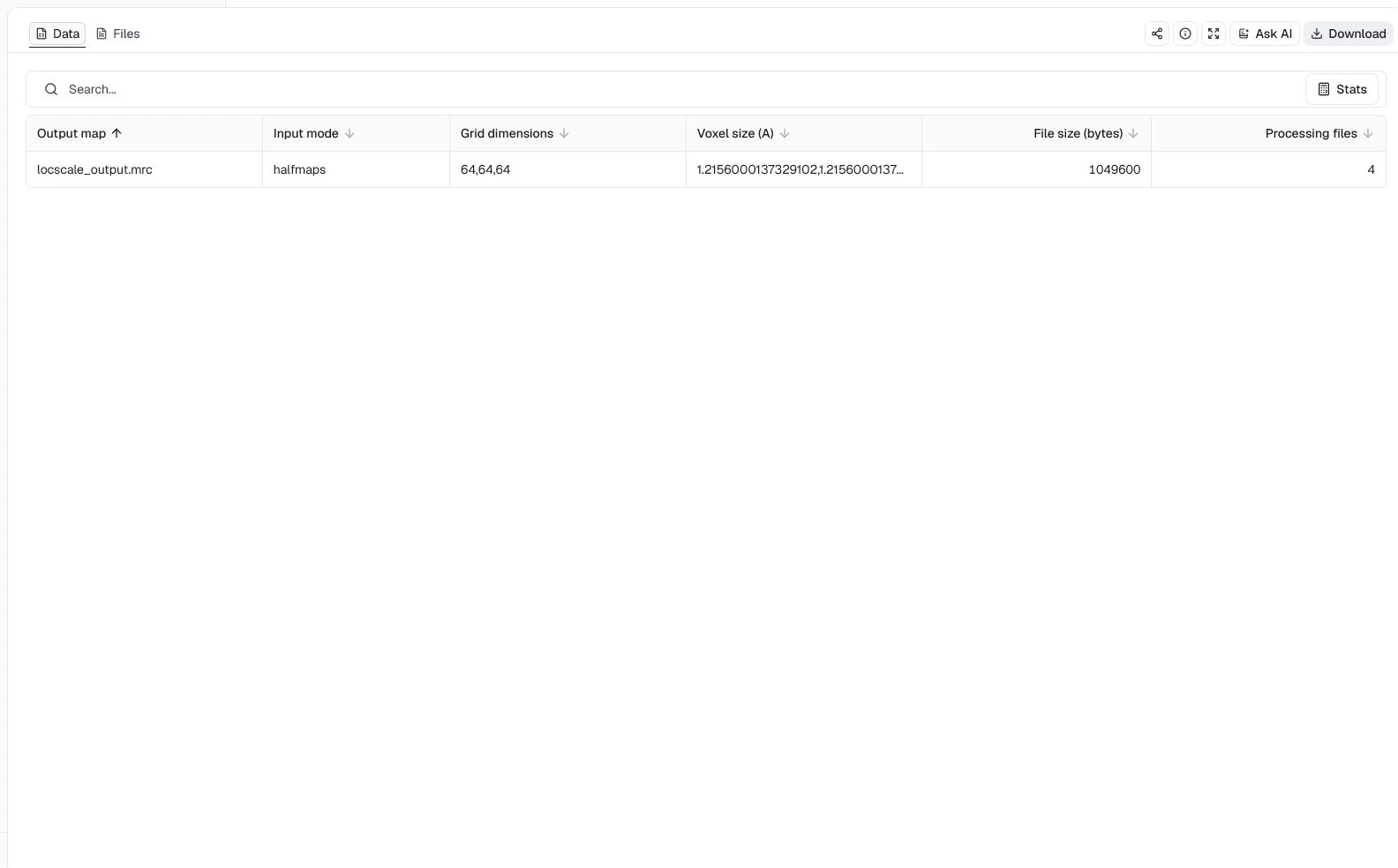
The completed job returns locscale_output.mrc on the original 64 × 64 × 64 grid, together with the reconstructed unfiltered map, bfactor_map.mrc, qfit_map.mrc, and the processing log. The table confirms that LocScale used halfmaps mode and preserved the input voxel spacing. This result shows that the paired-map and FSC-filtering workflow completed; it does not by itself establish improved interpretability. Compare the output with the input half-maps in a cryo-EM viewer before using it for model building.
This EMD-2984 example shows full-map operation on a high-resolution region of Escherichia coli β-galactosidase. The input is an average of matching deposited half-map subvolumes, accompanied by a data-derived mask and the corresponding deposited reconstruction as the direct scaling reference.
Map input = Full map, CPU processes = 4, Resolution cutoff (A) = 2.2, and Window size (pixels) = 16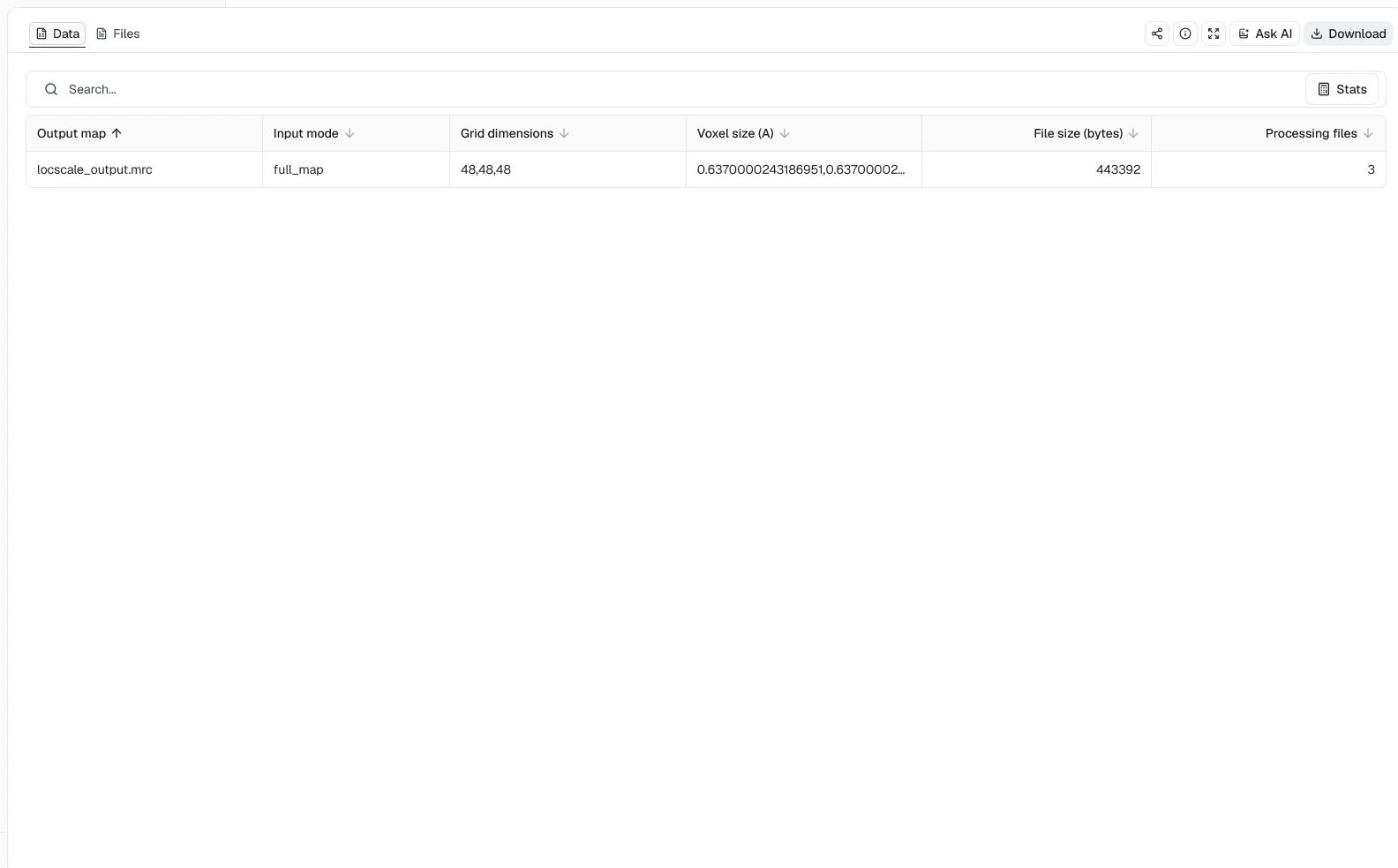
ProteinIQ returns a 48 × 48 × 48 sharpened MRC map at the same 0.637 Å voxel spacing, plus bfactor_map.mrc, qfit_map.mrc, and the LocScale log. The compact subvolume makes the reference-map workflow quick to inspect, but it is not a substitute for processing and evaluating the complete EMD-2984 reconstruction.
This example uses a compact region from the EMD-3061 human γ-secretase half-map pair to demonstrate full-map processing with both an explicit mask and input filtering. The supplied reference is the matching region from the deposited reconstruction.
Map input = Full map, CPU processes = 4, Resolution cutoff (A) = 3.4, Window size (pixels) = 24, and Filter input maps = enabled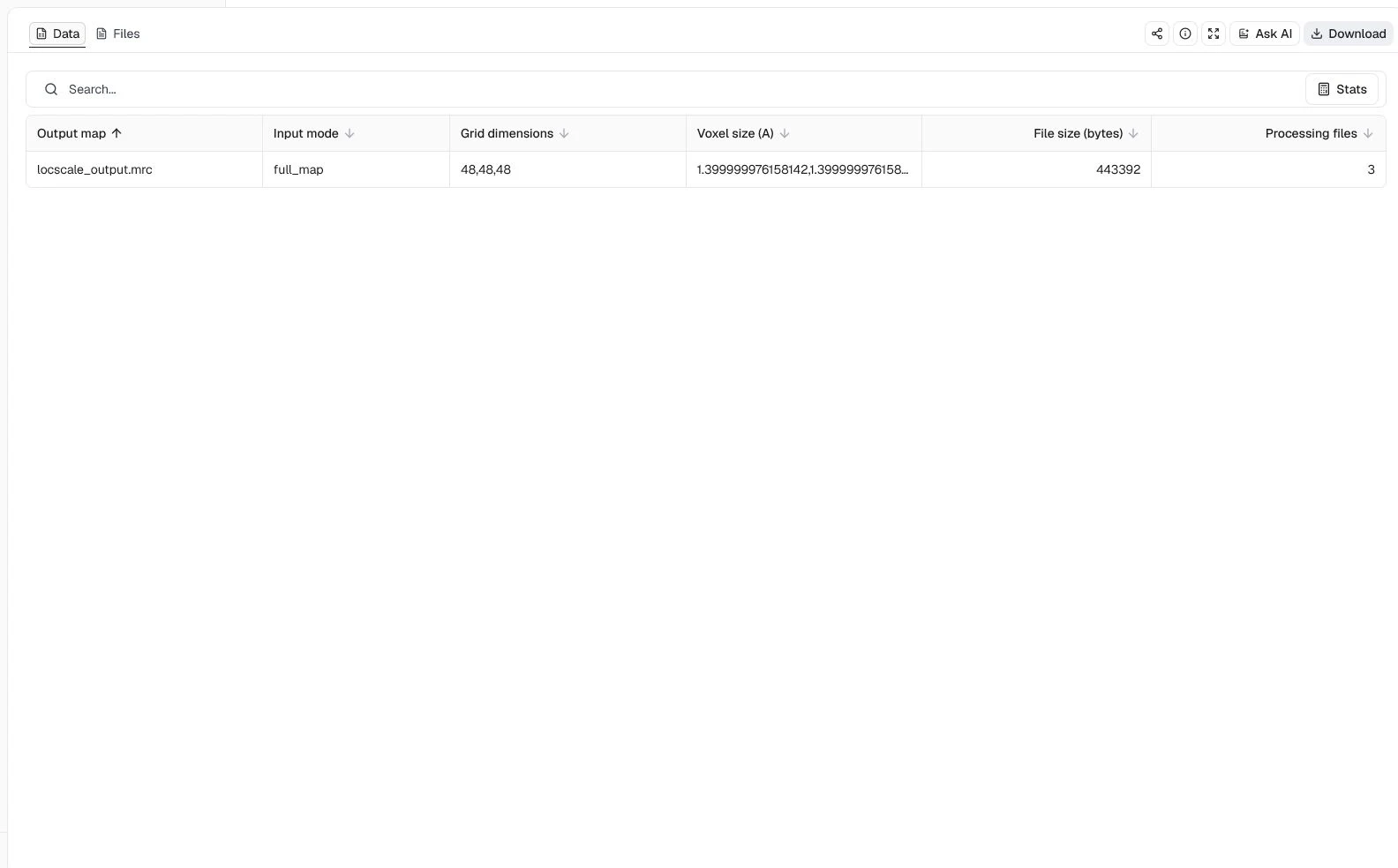
The output table records the expected 48 × 48 × 48 grid, 1.4 Å voxel spacing, and three processing files. This confirms that LocScale completed the masked, filtered full-map workflow and returned its sharpened map and diagnostics. Whether filtering improves local density for a particular structural interpretation still requires direct comparison with the unfiltered input and independent validation against the half-maps.
C1 until symmetry-specific runtime checks are added.