Validate generated and docked ligand poses with structure, chemistry, and reference-pose checks. Learn more
Run
5 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job”.
Validate generated and docked ligand poses with structure, chemistry, and reference-pose checks. Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
Validate generated and docked ligand poses with structure, chemistry, and reference-pose checks. Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
PoseBusters checks whether generated or docked ligand poses are chemically sane, geometrically plausible, and compatible with an optional protein binding site. The method was introduced by Buttenschoen, Morris, and Deane after showing that low RMSD alone can hide broken chemistry, impossible bond geometry, ligand self-clashes, and receptor overlaps in modern docking benchmarks.
The main question is not "did the pose score well?" It is "could this pose exist as a molecule in this pocket?" That makes PoseBusters useful after docking with GNINA, DiffDock, DynamicBind, SigmaDock, or other pose generation tools, especially when comparing many ranked conformations.
Run PoseBusters online by uploading a predicted ligand pose, optionally adding a reference ligand and protein receptor, then choosing the validation mode. ProteinIQ returns a downloadable table of pass/fail checks for ligand chemistry, 3D geometry, receptor clashes, and reference-pose agreement when reference inputs are supplied.
PoseBusters can run with just a ligand pose, but the most informative checks require the files that define the intended docking context.
| Input | Required | Accepted formats | Description |
|---|---|---|---|
Predicted/generated molecule | Yes | SDF, MOL, MOL2, PDB | One or more ligand poses or generated 3D molecules to validate. SDF is preferred for multi-pose files because conformers and molecule records are handled cleanly. |
True/reference ligand | No | SDF, MOL, MOL2, PDB | The crystallographic or trusted ligand pose used for identity, stereochemistry, and RMSD-style checks. The file should contain 3D coordinates. |
Conditioning protein/receptor | No | PDB, SDF, MOL, MOL2, RCSB PDB fetch | The receptor or protein structure used for protein-ligand distance, pocket placement, and volume-overlap checks. PDB is recommended for protein inputs because residue metadata helps classify protein atoms, cofactors, metals, and waters. |
Each predicted molecule file can contain several poses, and a job can include up to 20 predicted molecule files. PoseBusters reports one row per molecule or conformer unless Top poses to validate limits the run.
| Setting | Description |
|---|---|
Validation mode | Selects the PoseBusters check set. Auto-select from inputs chooses the appropriate mode from the provided predicted, reference, and receptor files. |
Output format | Controls the PoseBusters report style: Short, Long, or CSV. ProteinIQ displays the parsed table and keeps the results downloadable. |
Full report | Adds numeric diagnostic columns beyond pass/fail status, such as bond outlier counts, clash counts, ring planarity distances, and energy ratios. |
Top poses to validate | Restricts validation to the first N poses in the predicted molecule file. Leave blank to validate every pose. |
Auto-select from inputs is usually the safest choice. Manual modes are useful when the files are available but the intended evaluation task is narrower than the full input set.
| Mode | Best for | Inputs normally used | Main checks |
|---|---|---|---|
Molecule only | Generated conformers, molecule generators, or ligand cleanup QC | Predicted molecule | Loading, sanitization, InChI conversion, connectivity, radicals, bond lengths, bond angles, internal clashes, ring flatness, double-bond flatness, internal energy |
Dock | Docked ligands or newly generated ligands placed into a receptor pocket | Predicted molecule, receptor | Molecule-only checks plus protein-ligand distance and volume-overlap checks against protein atoms, cofactors, metals, and waters |
Redock | Re-docking benchmark cases where a crystallographic ligand is available | Predicted molecule, reference ligand, receptor | Dock checks plus molecular identity, bond/stereochemistry agreement, and RMSD within 2 Angstrom reference agreement |
Regenerate | Generated 3D conformers for a known molecule with a reference ligand | Predicted molecule, reference ligand | Molecule identity and geometry checks without receptor-pocket checks |
Generate | De novo molecule generation with reference-shape and receptor context | Predicted molecule, reference ligand, receptor | Chemistry, geometry, receptor compatibility, and SuCOS reference-shape/feature agreement without the molecular identity checks used by Redock or Regenerate |
Fast modes run the corresponding check set with speed-oriented settings. They are useful for triage on large SDF files, followed by the full mode on the best candidates.
The results table starts with file and molecule identifiers, then reports Boolean check columns. True means the pose passed that check. False marks a specific failure worth inspecting before using the pose for ranking, rescoring, molecular dynamics, or free-energy calculations.
| Column group | Common columns | Meaning |
|---|---|---|
| Loading | mol_pred_loaded, mol_true_loaded, mol_cond_loaded | Whether each supplied structure could be read. A failed loading check makes later checks unreliable or unavailable. |
| Chemical validity | sanitization, inchi_convertible, all_atoms_connected, no_radicals | RDKit-based checks for chemically valid molecule graphs. Failures often point to malformed files, disconnected fragments, invalid valence, radicals, or structures that need repair before docking analysis. |
| Identity agreement | molecular_formula, molecular_bonds, double_bond_stereochemistry, tetrahedral_chirality | Checks used when a reference ligand is supplied. Failures mean the predicted pose may not represent the same ligand, even if the coordinates look close. |
| Intramolecular geometry | bond_lengths, bond_angles, internal_steric_clash, aromatic_ring_flatness, non-aromatic_ring_non-flatness, double_bond_flatness, internal_energy | Tests for impossible or strained conformations inside the ligand. These catch poses with distorted bonds, atoms sitting on top of each other, twisted aromatic systems, or unusually high conformer energy. |
| Receptor placement | protein-ligand_maximum_distance, minimum_distance_to_protein, volume_overlap_with_protein, cofactor and water overlap columns | Checks whether the ligand is placed in a plausible protein environment rather than far from the pocket or clashing with receptor atoms. |
| Reference agreement | RMSD within 2 Angstrom | Indicates whether the predicted ligand pose is within the common 2 Angstrom RMSD success criterion when a reference ligand is available. |
Full report adds diagnostic measurements that explain failures. For example, number_clashes helps separate one local contact from a badly collapsed conformer, while energy_ratio highlights conformations that pass simple bond checks but remain energetically strained.
A good docking result should pass both physical plausibility checks and task-specific accuracy checks. In redocking, a pose within 2 Angstrom RMSD but with failed stereochemistry or volume overlap should not be counted as a clean success. The pose may be close to the crystal ligand by coordinate RMSD while still representing the wrong stereoisomer or colliding with protein atoms.
Failures have different practical meanings:
Passing PoseBusters does not prove that a pose binds strongly. It proves that the pose clears a set of chemistry and geometry sanity checks. Binding affinity, ranking, induced fit, waters, protonation states, and assay relevance remain separate questions.
PoseBusters runs a suite of RDKit-based tests against the predicted ligand and any optional context structures. The checks combine molecule graph validation, 3D geometry validation, receptor-contact analysis, and optional reference comparison.
The paper emphasized a practical benchmarking problem: many AI docking methods were evaluated mainly by ligand RMSD, but some low-RMSD poses contained nonphysical chemistry. PoseBusters adds a second axis to pose evaluation. A method should recover native-like binding modes and generate structures that pass basic chemistry and steric criteria.
The check set changes with the validation mode:
PoseBusters is stricter than a visual inspection because it catches subtle graph and geometry problems that can be hard to see in a 3D viewer. It is also narrower than a physics simulation: it does not relax the pose, compute binding free energy, model receptor flexibility, or decide whether the ligand is a true binder.
| Tool | Best question | Typical use |
|---|---|---|
| PoseBusters | Is this ligand pose chemically and geometrically plausible? | Quality control after docking, molecule generation, or benchmarking. |
| MolProbity | Is this protein structure geometrically sound? | Protein model validation, clashscore, rotamer, Ramachandran, and all-atom contact checks. |
| DockQ | How close is a predicted protein complex to a reference complex? | Protein-protein docking benchmark evaluation. |
| GNINA | What ligand pose and CNN score does a docking model predict? | Protein-ligand docking and CNN rescoring before PoseBusters validation. |
| DiffDock | What poses does a diffusion docking model generate quickly? | Generating candidate ligand poses that can then be checked with PoseBusters. |
| SurfDock | What poses does a surface-aware diffusion docking model generate? | Generating ligand poses with surface-aware geometric context before validation. |
| PDB Fixer | Is the receptor structure ready for downstream modeling? | Adding missing atoms, repairing residues, and preparing PDB structures before docking or validation. |
For ligand-pose workflows, a common sequence is docking first, PoseBusters second, and deeper analysis third. For example, candidate poses from GNINA or DiffDock can be filtered for chemistry and receptor clashes before expensive molecular dynamics or free-energy calculations with GROMACS or OpenFE.
PoseBusters results depend on the structure files supplied. Missing hydrogens, unusual protonation, metal coordination, alternate locations, covalent ligands, or incomplete receptor preparation can create failures that need chemical review rather than automatic rejection.
For benchmark reporting, keep the mode consistent across methods. Comparing one docking method with Redock checks and another with Molecule only checks mixes different definitions of success. Report both the physical pass rate and the pose-recovery metric when a reference ligand is available.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.

Analyze noncovalent interactions in protein-ligand complex structures with PLIP, including hydrogen bonds, hydrophobic contacts, pi interactions, salt bridges, water bridges, halogen bonds, and metal complexes.
ProLIF calculates protein-ligand interaction fingerprints from 3D structures, returning residue-level interaction tables, interaction metadata, and native fingerprint files.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Calculate pairwise RMSD matrices for PDB structure ensembles with pyRMSD, including the condensed matrix and source statistics files.
Calculate RMSD between protein structures with optimal rigid-body alignment, explicit atom correspondence, coverage diagnostics, per-residue results, and fitted coordinate files.
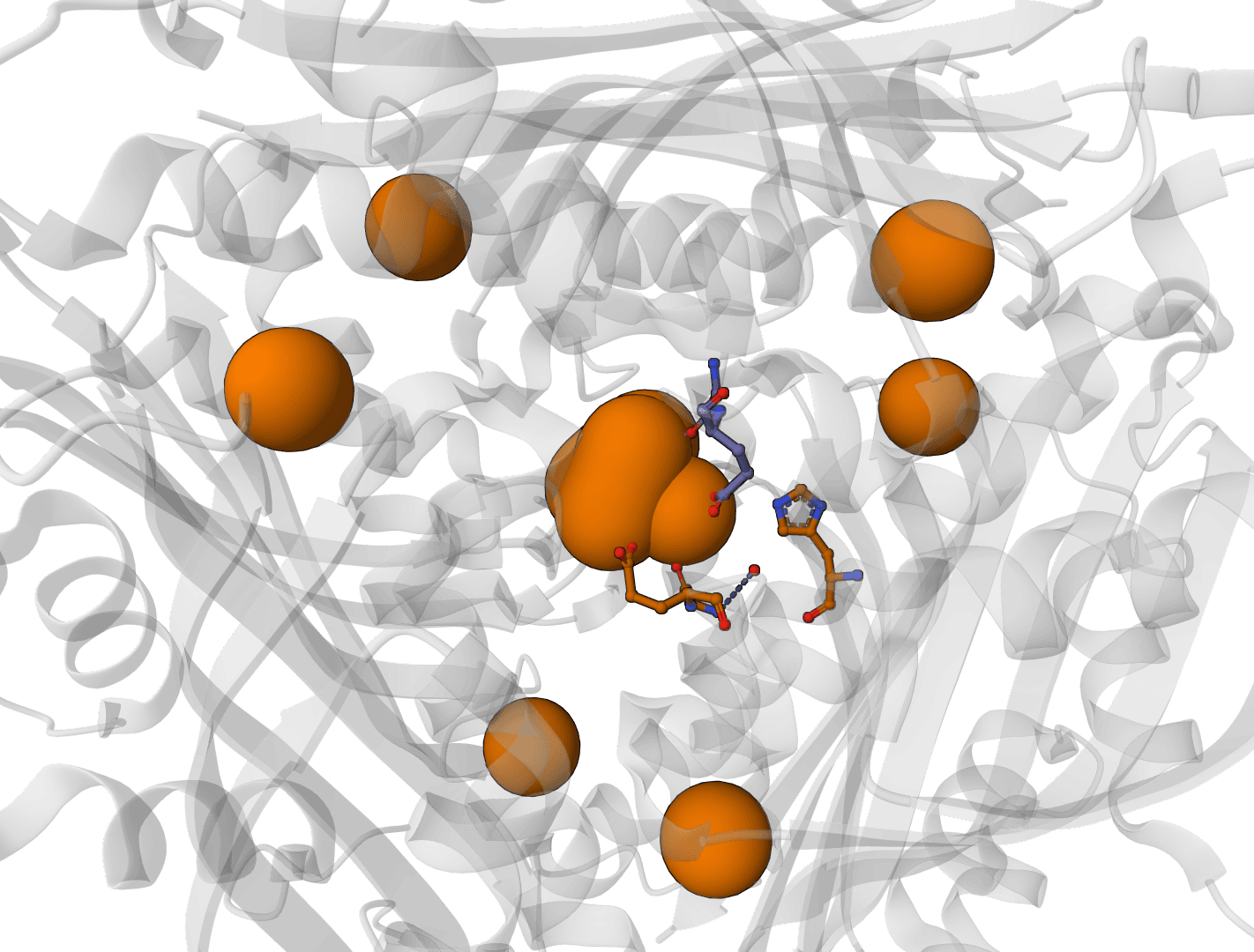
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.
PoseBusters checks whether generated or docked ligand poses are chemically sane, geometrically plausible, and compatible with an optional protein binding site. The method was introduced by Buttenschoen, Morris, and Deane after showing that low RMSD alone can hide broken chemistry, impossible bond geometry, ligand self-clashes, and receptor overlaps in modern docking benchmarks.
The main question is not "did the pose score well?" It is "could this pose exist as a molecule in this pocket?" That makes PoseBusters useful after docking with GNINA, DiffDock, DynamicBind, SigmaDock, or other pose generation tools, especially when comparing many ranked conformations.
Run PoseBusters online by uploading a predicted ligand pose, optionally adding a reference ligand and protein receptor, then choosing the validation mode. ProteinIQ returns a downloadable table of pass/fail checks for ligand chemistry, 3D geometry, receptor clashes, and reference-pose agreement when reference inputs are supplied.
PoseBusters can run with just a ligand pose, but the most informative checks require the files that define the intended docking context.
| Input | Required | Accepted formats | Description |
|---|---|---|---|
Predicted/generated molecule | Yes | SDF, MOL, MOL2, PDB | One or more ligand poses or generated 3D molecules to validate. SDF is preferred for multi-pose files because conformers and molecule records are handled cleanly. |
True/reference ligand | No | SDF, MOL, MOL2, PDB | The crystallographic or trusted ligand pose used for identity, stereochemistry, and RMSD-style checks. The file should contain 3D coordinates. |
Conditioning protein/receptor | No | PDB, SDF, MOL, MOL2, RCSB PDB fetch | The receptor or protein structure used for protein-ligand distance, pocket placement, and volume-overlap checks. PDB is recommended for protein inputs because residue metadata helps classify protein atoms, cofactors, metals, and waters. |
Each predicted molecule file can contain several poses, and a job can include up to 20 predicted molecule files. PoseBusters reports one row per molecule or conformer unless Top poses to validate limits the run.
| Setting | Description |
|---|---|
Validation mode | Selects the PoseBusters check set. Auto-select from inputs chooses the appropriate mode from the provided predicted, reference, and receptor files. |
Output format | Controls the PoseBusters report style: Short, Long, or CSV. ProteinIQ displays the parsed table and keeps the results downloadable. |
Full report | Adds numeric diagnostic columns beyond pass/fail status, such as bond outlier counts, clash counts, ring planarity distances, and energy ratios. |
Top poses to validate | Restricts validation to the first N poses in the predicted molecule file. Leave blank to validate every pose. |
Auto-select from inputs is usually the safest choice. Manual modes are useful when the files are available but the intended evaluation task is narrower than the full input set.
| Mode | Best for | Inputs normally used | Main checks |
|---|---|---|---|
Molecule only | Generated conformers, molecule generators, or ligand cleanup QC | Predicted molecule | Loading, sanitization, InChI conversion, connectivity, radicals, bond lengths, bond angles, internal clashes, ring flatness, double-bond flatness, internal energy |
Dock | Docked ligands or newly generated ligands placed into a receptor pocket | Predicted molecule, receptor | Molecule-only checks plus protein-ligand distance and volume-overlap checks against protein atoms, cofactors, metals, and waters |
Redock | Re-docking benchmark cases where a crystallographic ligand is available | Predicted molecule, reference ligand, receptor | Dock checks plus molecular identity, bond/stereochemistry agreement, and RMSD within 2 Angstrom reference agreement |
Regenerate | Generated 3D conformers for a known molecule with a reference ligand | Predicted molecule, reference ligand | Molecule identity and geometry checks without receptor-pocket checks |
Generate | De novo molecule generation with reference-shape and receptor context | Predicted molecule, reference ligand, receptor | Chemistry, geometry, receptor compatibility, and SuCOS reference-shape/feature agreement without the molecular identity checks used by Redock or Regenerate |
Fast modes run the corresponding check set with speed-oriented settings. They are useful for triage on large SDF files, followed by the full mode on the best candidates.
The results table starts with file and molecule identifiers, then reports Boolean check columns. True means the pose passed that check. False marks a specific failure worth inspecting before using the pose for ranking, rescoring, molecular dynamics, or free-energy calculations.
| Column group | Common columns | Meaning |
|---|---|---|
| Loading | mol_pred_loaded, mol_true_loaded, mol_cond_loaded | Whether each supplied structure could be read. A failed loading check makes later checks unreliable or unavailable. |
| Chemical validity | sanitization, inchi_convertible, all_atoms_connected, no_radicals | RDKit-based checks for chemically valid molecule graphs. Failures often point to malformed files, disconnected fragments, invalid valence, radicals, or structures that need repair before docking analysis. |
| Identity agreement | molecular_formula, molecular_bonds, double_bond_stereochemistry, tetrahedral_chirality | Checks used when a reference ligand is supplied. Failures mean the predicted pose may not represent the same ligand, even if the coordinates look close. |
| Intramolecular geometry | bond_lengths, bond_angles, internal_steric_clash, aromatic_ring_flatness, non-aromatic_ring_non-flatness, double_bond_flatness, internal_energy | Tests for impossible or strained conformations inside the ligand. These catch poses with distorted bonds, atoms sitting on top of each other, twisted aromatic systems, or unusually high conformer energy. |
| Receptor placement | protein-ligand_maximum_distance, minimum_distance_to_protein, volume_overlap_with_protein, cofactor and water overlap columns | Checks whether the ligand is placed in a plausible protein environment rather than far from the pocket or clashing with receptor atoms. |
| Reference agreement | RMSD within 2 Angstrom | Indicates whether the predicted ligand pose is within the common 2 Angstrom RMSD success criterion when a reference ligand is available. |
Full report adds diagnostic measurements that explain failures. For example, number_clashes helps separate one local contact from a badly collapsed conformer, while energy_ratio highlights conformations that pass simple bond checks but remain energetically strained.
A good docking result should pass both physical plausibility checks and task-specific accuracy checks. In redocking, a pose within 2 Angstrom RMSD but with failed stereochemistry or volume overlap should not be counted as a clean success. The pose may be close to the crystal ligand by coordinate RMSD while still representing the wrong stereoisomer or colliding with protein atoms.
Failures have different practical meanings:
Passing PoseBusters does not prove that a pose binds strongly. It proves that the pose clears a set of chemistry and geometry sanity checks. Binding affinity, ranking, induced fit, waters, protonation states, and assay relevance remain separate questions.
PoseBusters runs a suite of RDKit-based tests against the predicted ligand and any optional context structures. The checks combine molecule graph validation, 3D geometry validation, receptor-contact analysis, and optional reference comparison.
The paper emphasized a practical benchmarking problem: many AI docking methods were evaluated mainly by ligand RMSD, but some low-RMSD poses contained nonphysical chemistry. PoseBusters adds a second axis to pose evaluation. A method should recover native-like binding modes and generate structures that pass basic chemistry and steric criteria.
The check set changes with the validation mode:
PoseBusters is stricter than a visual inspection because it catches subtle graph and geometry problems that can be hard to see in a 3D viewer. It is also narrower than a physics simulation: it does not relax the pose, compute binding free energy, model receptor flexibility, or decide whether the ligand is a true binder.
| Tool | Best question | Typical use |
|---|---|---|
| PoseBusters | Is this ligand pose chemically and geometrically plausible? | Quality control after docking, molecule generation, or benchmarking. |
| MolProbity | Is this protein structure geometrically sound? | Protein model validation, clashscore, rotamer, Ramachandran, and all-atom contact checks. |
| DockQ | How close is a predicted protein complex to a reference complex? | Protein-protein docking benchmark evaluation. |
| GNINA | What ligand pose and CNN score does a docking model predict? | Protein-ligand docking and CNN rescoring before PoseBusters validation. |
| DiffDock | What poses does a diffusion docking model generate quickly? | Generating candidate ligand poses that can then be checked with PoseBusters. |
| SurfDock | What poses does a surface-aware diffusion docking model generate? | Generating ligand poses with surface-aware geometric context before validation. |
| PDB Fixer | Is the receptor structure ready for downstream modeling? | Adding missing atoms, repairing residues, and preparing PDB structures before docking or validation. |
For ligand-pose workflows, a common sequence is docking first, PoseBusters second, and deeper analysis third. For example, candidate poses from GNINA or DiffDock can be filtered for chemistry and receptor clashes before expensive molecular dynamics or free-energy calculations with GROMACS or OpenFE.
PoseBusters results depend on the structure files supplied. Missing hydrogens, unusual protonation, metal coordination, alternate locations, covalent ligands, or incomplete receptor preparation can create failures that need chemical review rather than automatic rejection.
For benchmark reporting, keep the mode consistent across methods. Comparing one docking method with Redock checks and another with Molecule only checks mixes different definitions of success. Report both the physical pass rate and the pose-recovery metric when a reference ligand is available.
Assess docking model quality by comparing predicted complexes against native references. DockQ v2.1.3 supports protein, nucleic-acid, and supported small-molecule interfaces with faithful native metrics.
Analyze noncovalent interactions in protein-ligand complex structures with PLIP, including hydrogen bonds, hydrophobic contacts, pi interactions, salt bridges, water bridges, halogen bonds, and metal complexes.
ProLIF calculates protein-ligand interaction fingerprints from 3D structures, returning residue-level interaction tables, interaction metadata, and native fingerprint files.
Scoring function for interprotein interactions in AlphaFold2, AlphaFold3 and Boltz predictions. Calculates ipSAE, ipTM, pDockQ, pDockQ2, and LIS scores to assess protein-protein interface quality.
Validate protein structure quality with all-atom contact analysis, Ramachandran plots, rotamer assessment, and geometry checks.
Generate a downloadable PDBsum structural summary report archive for a single protein structure.
Calculate pairwise RMSD matrices for PDB structure ensembles with pyRMSD, including the condensed matrix and source statistics files.
Calculate RMSD between protein structures with optimal rigid-body alignment, explicit atom correspondence, coverage diagnostics, per-residue results, and fitted coordinate files.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Assign protein secondary structure using the DSSP algorithm. The gold standard for hydrogen bond-based structure assignment from coordinates.