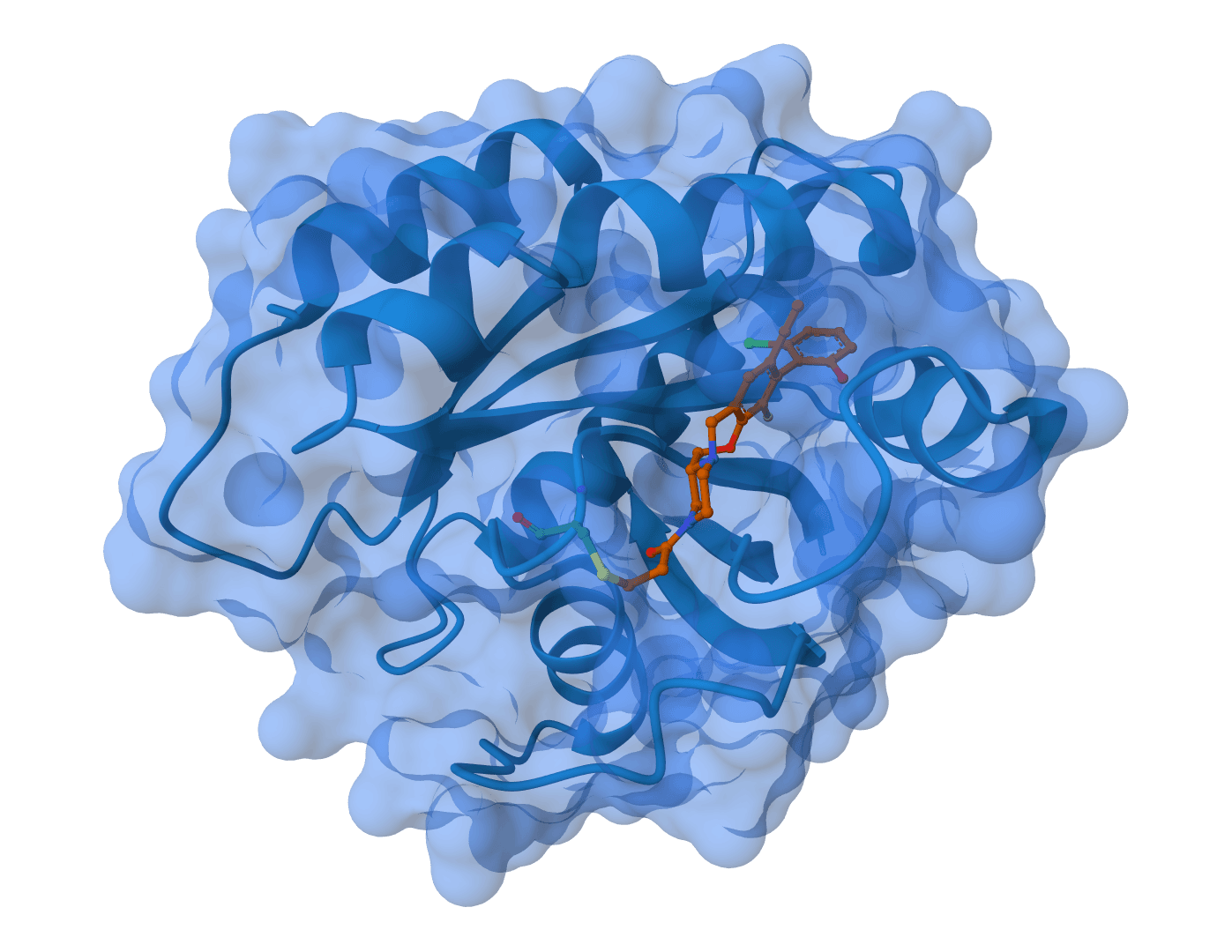
OpenFE (Open Free Energy) is an open-source Python framework for alchemical free energy calculations, a physics-based computational method used in drug discovery to predict how strongly molecules interact with their environment or a protein target. Rather than scoring a single static pose like molecular docking, alchemical methods simulate the thermodynamics of molecular transformations, yielding quantitative binding affinity estimates in kcal/mol with statistical uncertainty.
The framework supports two main calculation types:
Absolute Hydration Free Energy (AHFE): The free energy change when a molecule is transferred from vacuum into water. AHFE quantifies how much a compound prefers aqueous solution over the gas phase, a property directly linked to solubility and membrane permeability.
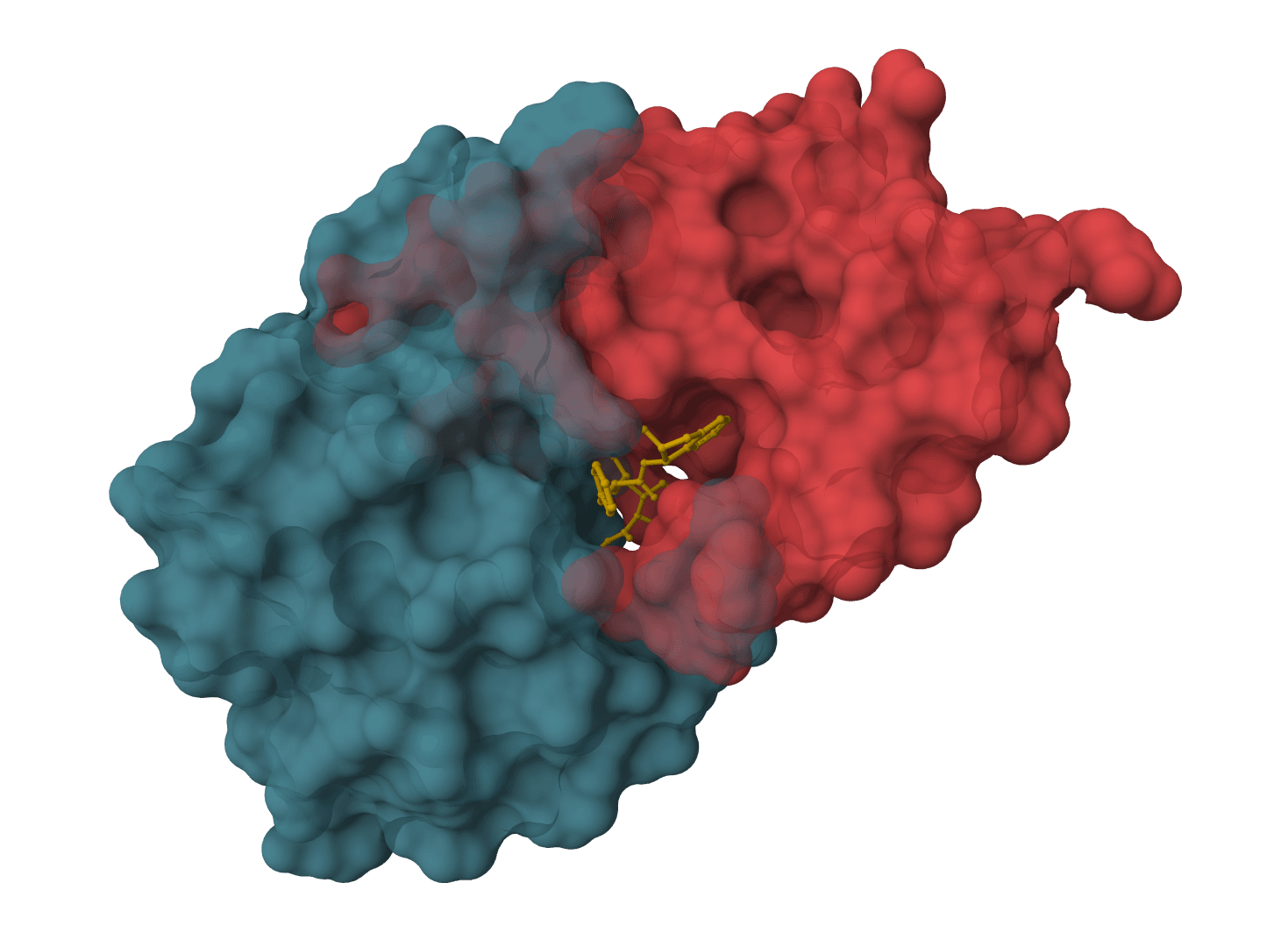
Relative Binding Free Energy (RBFE): The difference in binding affinity between two ligands at a protein target. RBFE is the workhorse of free energy methods in pharmaceutical lead optimization, predicting which chemical modifications improve or weaken binding without synthesizing every candidate.
OpenFE is developed by a consortium of pharmaceutical companies and academic groups, with GPU-accelerated simulations powered by OpenMM.
How alchemical free energy calculations work
In classical thermodynamics, measuring a binding free energy directly would require simulating the full association and dissociation of a ligand, an event that happens on timescales far beyond what molecular dynamics can reach. Alchemical methods sidestep this by exploiting the fact that free energy is a state function: the path between two states does not matter, only the endpoints.
Instead of physically pulling a ligand out of a binding pocket, the calculation gradually "switches off" the ligand's interactions with its surroundings through a series of unphysical intermediate states. A coupling parameter λ controls this transformation, varying from 0 (full interactions) to 1 (fully decoupled). At each λ value, a short molecular dynamics simulation samples the local thermodynamics.
Lambda windows
The number of λ intermediates is called the lambda window count. More windows provide smoother overlap between adjacent states and more reliable free energy estimates, at the cost of additional simulation time. A typical AHFE calculation uses 11-14 windows; RBFE calculations may need more depending on the size of the chemical transformation.
AHFE thermodynamic cycle
For hydration free energies, the ligand is decoupled in two environments independently:
In solvent: electrostatic interactions are annihilated, then van der Waals interactions are decoupled
In vacuum: the same decoupling is performed without solvent
The hydration free energy is the difference: ΔGhydration=ΔGsolvent−ΔGvacuum.
RBFE thermodynamic cycle
For relative binding free energies, ligand A is alchemically transformed into ligand B in two legs:
In complex: A is morphed into B while bound to the protein
In solvent: the same A-to-B transformation in water alone
The relative binding free energy is: ΔΔGbind=ΔGcomplex−ΔGsolvent.
Both legs require the ligands to share a common molecular scaffold so that the alchemical transformation is tractable.
Analysis
Free energies are extracted from the simulation data using MBAR (Multistate Bennett Acceptance Ratio), which simultaneously analyzes energy differences across all λ windows for statistically optimal estimates.
How to use OpenFE online
ProteinIQ provides cloud-hosted OpenFE calculations on GPU infrastructure, handling all environment setup, force field parameterization, and simulation orchestration automatically.
Inputs
Input
Description
Protein Structure
PDB file, mmCIF file, or RCSB PDB ID (e.g., 3HTB). Required for RBFE calculations, not used for AHFE.
Ligand
SMILES string or SDF/MOL/MOL2 file describing the small molecule.
For AHFE calculations, only the ligand is needed. For RBFE, both protein structure and ligand are required.
Calculation settings
Setting
Description
Calculation Type
Absolute Hydration Free Energy runs an AHFE calculation (ligand in water vs vacuum). Relative Binding Free Energy runs an RBFE calculation against a protein target.
Simulation Length
Duration per lambda window. Short (0.5 ns) for quick estimates, Medium (2 ns) for reasonable convergence, Long (10 ns) for publication-quality results.
Number of Repeats
Independent repeat calculations. 1 is faster; 3 is recommended for reliable uncertainty estimates.
Advanced settings
Setting
Description
Lambda windows
Number of alchemical intermediates (5-24, default 11). More windows improve accuracy at increased cost.
Small molecule force field
OpenFF Sage 2.2.0 (recommended), OpenFF Sage 2.1.0, or GAFF 2.11 for ligand parameterization.
Water model
TIP3P (fast, well-validated) or TIP4P-Ew (more accurate solvation thermodynamics, slower).
Results
Output is a table of free energy estimates in kcal/mol with associated uncertainties. For AHFE, the result is the hydration free energy. For RBFE, the result is the relative binding free energy difference between the ligand and a reference state.
Interpreting results
AHFE values
Hydration free energies are typically negative for polar, water-soluble compounds and near zero or positive for hydrophobic molecules. Experimental AHFE values for druglike molecules generally range from +2 to −15 kcal/mol. Well-converged OpenFE AHFE calculations typically achieve accuracy within 1 kcal/mol of experimental values.
RBFE values
A negative ΔΔG indicates the ligand binds more strongly than the reference compound. In lead optimization, differences of 1-2 kcal/mol correspond to roughly 10-fold changes in binding affinity, significant enough to guide compound selection.
ΔΔG (kcal/mol)
Interpretation
< −2
Substantially stronger binding than reference
−1 to −2
Moderate improvement (~10-100x affinity gain)
−0.5 to 0.5
Similar affinity to reference
Uncertainty and convergence
Results include statistical uncertainty from MBAR analysis. If uncertainty exceeds 1 kcal/mol, the calculation may not be converged. Increasing simulation length, adding lambda windows, or running more repeats can improve convergence.
Limitations
RBFE requires a common scaffold: The two ligands being compared must share a core structure. Large R-group transformations or scaffold hops may fail or produce unreliable results.
Protein flexibility is limited: While sidechains near the binding site can rearrange during simulation, large-scale conformational changes (loop movements, domain motions) may not be captured within typical simulation timescales.
Force field dependence: Results are only as accurate as the underlying force field. Molecules with unusual functional groups may be poorly parameterized.
Convergence is not guaranteed: Short simulations with few lambda windows can produce results with small reported uncertainties that are nonetheless systematically wrong. When accuracy matters, use longer simulations with 3 repeats.