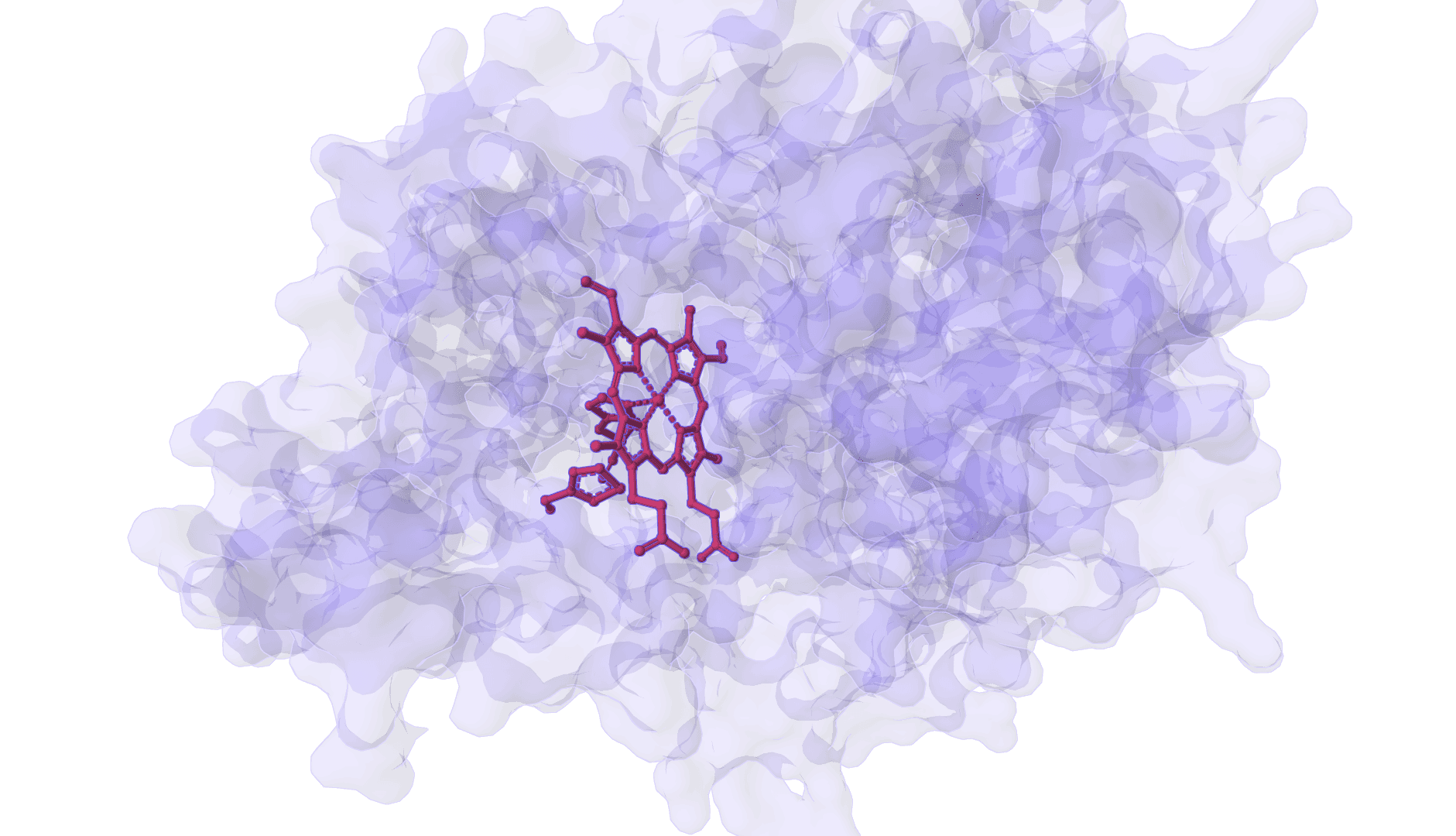
Predict protein-ligand binding poses while modeling ligand-induced conformational changes. Learn more
Run
50 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Simple binding example
Predict protein-ligand binding poses while modeling ligand-induced conformational changes. Learn more
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Simple binding example
Predict protein-ligand binding poses while modeling ligand-induced conformational changes. Learn more
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Simple binding example
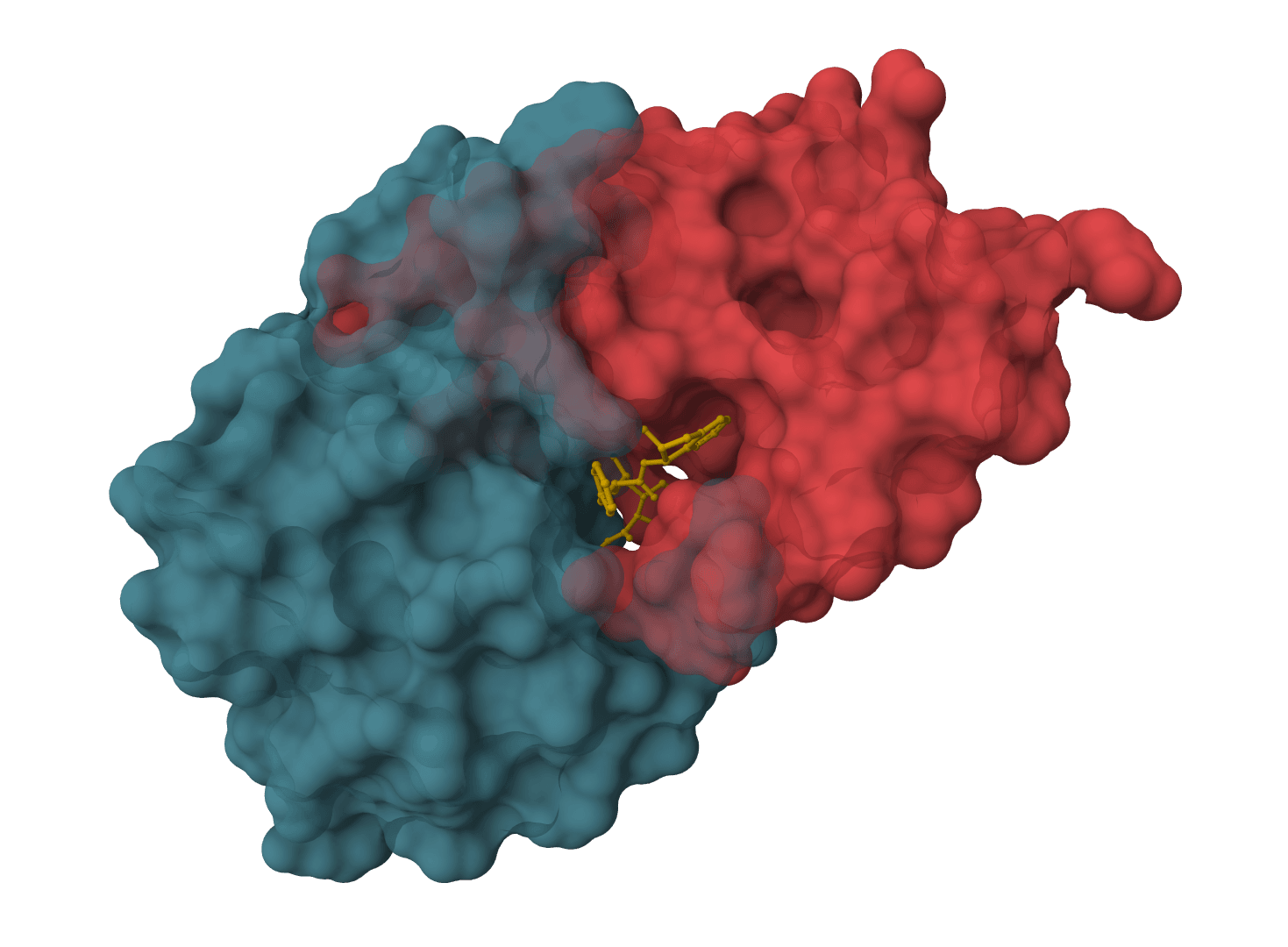
DynamicBind is a deep learning docking tool that predicts how small-molecule ligands bind to proteins while accounting for protein flexibility. Unlike traditional docking methods that treat proteins as rigid structures, DynamicBind predicts ligand-induced conformational changes—the structural adjustments proteins undergo when binding occurs.
This flexibility is critical for drug discovery. Proteins in their unbound (apo) state often have different side-chain orientations and backbone conformations compared to their ligand-bound (holo) state. DynamicBind recovers these holo-like conformations directly from apo structures, including AlphaFold predictions, without requiring prior knowledge of the binding site.
For traditional rigid docking approaches, see DiffDock, AutoDock Vina, or GNINA. For structure-based ligand generation, explore PocketFlow.
DynamicBind uses a diffusion-based generative model that learns to transition proteins from their apo state to ligand-specific holo conformations. The model is E(3)-equivariant, meaning it respects the 3D rotational and translational symmetries of molecular structures. This mathematical property allows the model to train on 1000× less data while achieving superior generalization compared to non-equivariant architectures.
The diffusion process operates over 20 inference steps with progressively smaller perturbations. Rather than adding random Gaussian noise, DynamicBind learns biologically relevant conformational transitions by training on paired apo-holo crystal structures from the PDBbind2020 dataset (19,443 protein-ligand complexes).
Each protein residue is represented as a node with two geometric features: backbone coordinates and side-chain dihedral angles ( angles). This coarse-graining reduces degrees of freedom while maintaining the ability to reconstruct all non-hydrogen atom positions. The model simultaneously predicts:
The use of tensor products of irreducible representations enables the model to capture complex protein-ligand interactions while preserving SE(3) equivariance throughout the neural network layers.
DynamicBind accommodates large protein conformational changes, including kinase DFG-in/out transitions and cryptic pocket opening. By learning smooth energy landscapes between equilibrium states, the model can recover holo-structures even when the initial AlphaFold prediction has large pocket RMSD ( Å) relative to the bound state.
The model outputs both the predicted ligand pose and the corresponding protein conformation, allowing users to visualize how binding site residues adjust to accommodate the ligand.
DynamicBind automatically removes water molecules, non-standard residues, and ligands during preprocessing. For structure prediction, use ESMFold, Boltz-2, or Chai-1.
The ligand is automatically processed and converted to the appropriate internal representation for docking.
Controls how many binding poses to generate per protein-ligand pair. More poses increase the chance of sampling near-native conformations but require longer computation time.
We recommend starting with 10 poses for initial exploration and increasing to 20-40 for final predictions or challenging targets where the binding site is unknown.
The number of diffusion denoising steps used during sampling. Higher values may improve pose quality by allowing more gradual refinement but increase computation time proportionally.
The model was trained and validated with 20 steps, which provides a good balance between quality and speed. Values below 15 may produce lower-quality poses, while values above 25 show diminishing returns.
When enabled, DynamicBind predicts ligand-induced conformational changes in both backbone and side-chain positions. When disabled, the model performs rigid docking with the input structure held fixed.
Enable flexibility (default) when working with apo structures or AlphaFold predictions. Disable for holo structures where the binding site conformation is already optimized, or when you want to test if a ligand can bind without requiring protein adjustments.
DynamicBind returns ranked binding poses with two scoring metrics:
Predicted binding affinity in units, where . Higher values indicate stronger binding:
The affinity prediction is learned from PDBbind experimental binding data and provides an estimate of binding strength. Note that affinity predictions are less reliable for proteins with low sequence homology to the training set.
Confidence metric ranging from 0 to 1 that estimates the accuracy of the predicted protein conformation. Higher lDDT scores indicate the model has high confidence in both the ligand pose and the induced protein conformational change.
Poses are ranked by a combination of lDDT and clash scores to prioritize geometrically plausible structures with favorable predicted confidence.
The top-ranked pose represents the model's best prediction, but examining multiple poses is recommended. For successful predictions on benchmark datasets:
Download predicted structures in SDF format (ligand) and PDB format (protein conformation) for further analysis or molecular dynamics simulations.
DynamicBind is particularly valuable when:
For these applications, DynamicBind achieves 1.7× higher success rates compared to DiffDock under stringent accuracy criteria.
Performance depends on similarity to the PDBbind2020 training set. Predictions for proteins with low sequence homology to known structures show reduced accuracy. The model performs best on major drug target families (kinases, GPCRs, nuclear receptors, ion channels) that are well-represented in crystallographic databases.
When the binding site is unknown, DynamicBind may struggle to identify the correct pocket, especially for novel protein targets. Providing a general binding region or using multiple inference runs with different random seeds can improve coverage.
Perfect pose selection using confidence scores could improve success rates from 33% to 50%, indicating that the current lDDT-based ranking occasionally prioritizes incorrect poses. Manual inspection of top-ranked poses is recommended for critical applications.
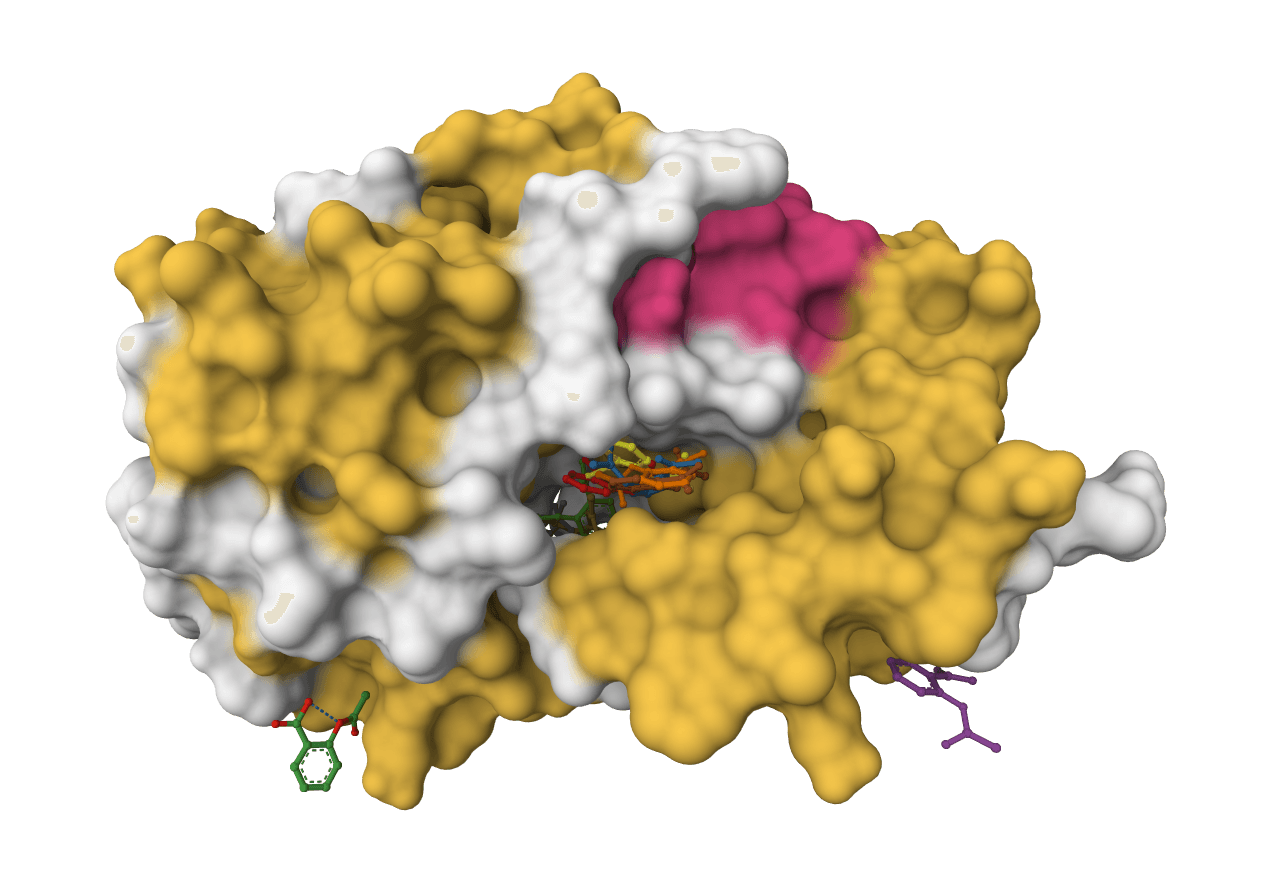
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
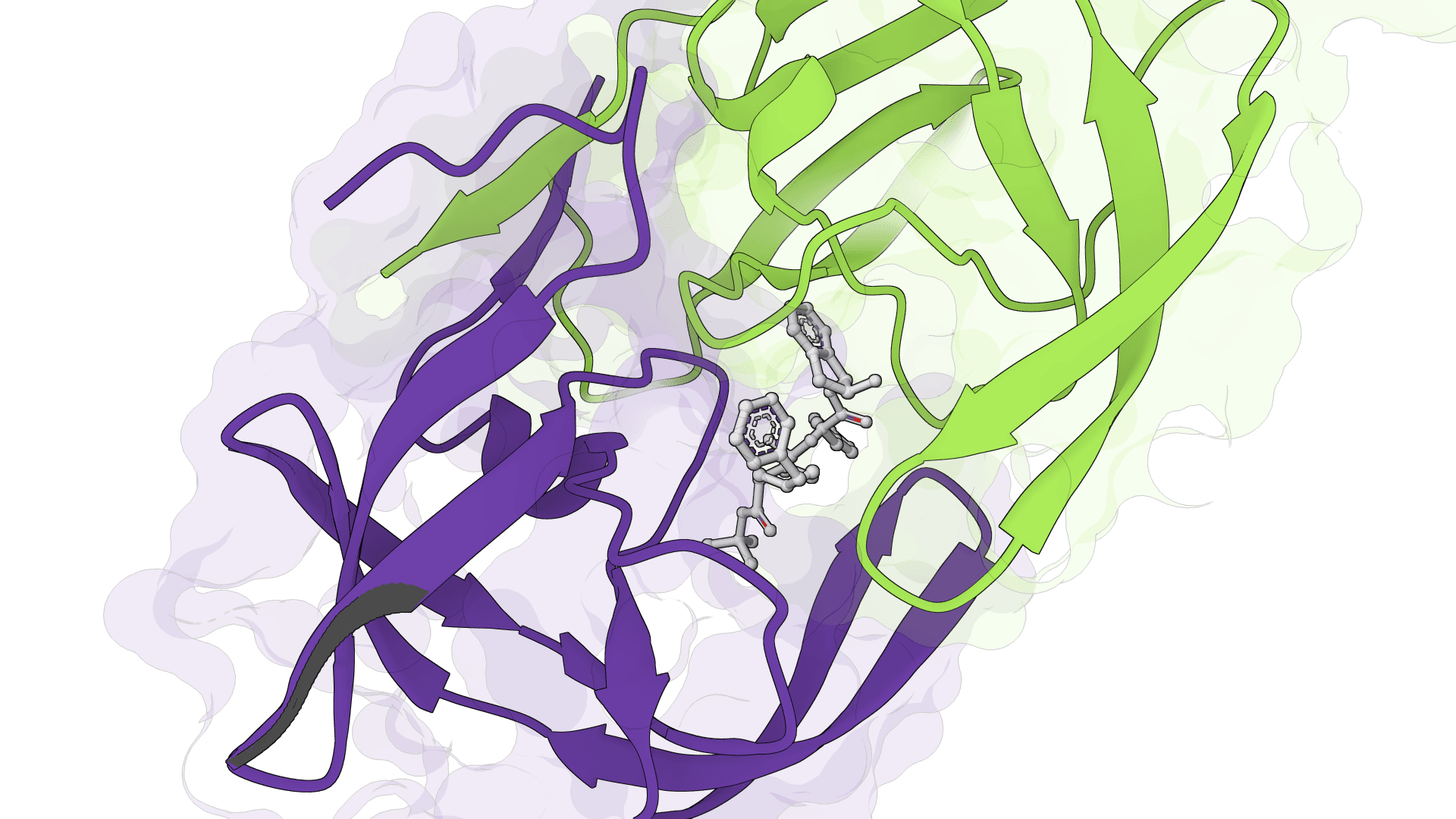
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
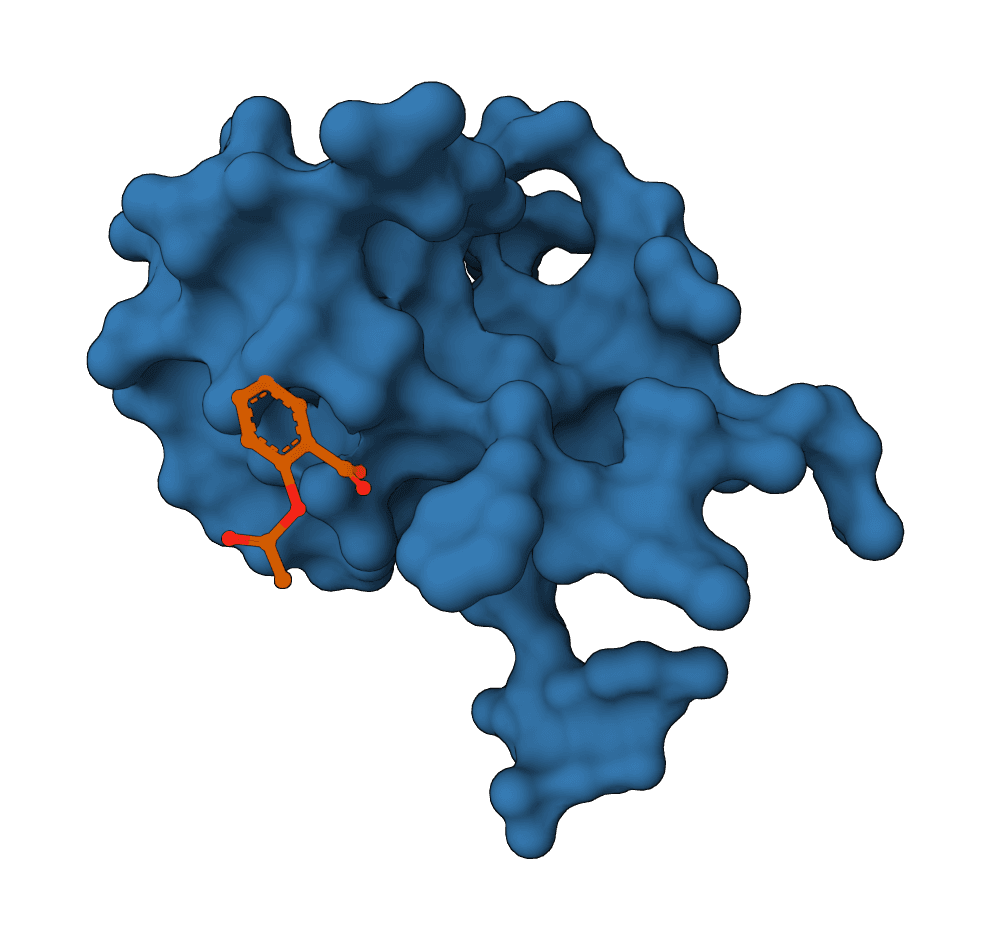
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
DynamicBind is a deep learning docking tool that predicts how small-molecule ligands bind to proteins while accounting for protein flexibility. Unlike traditional docking methods that treat proteins as rigid structures, DynamicBind predicts ligand-induced conformational changes—the structural adjustments proteins undergo when binding occurs.
This flexibility is critical for drug discovery. Proteins in their unbound (apo) state often have different side-chain orientations and backbone conformations compared to their ligand-bound (holo) state. DynamicBind recovers these holo-like conformations directly from apo structures, including AlphaFold predictions, without requiring prior knowledge of the binding site.
For traditional rigid docking approaches, see DiffDock, AutoDock Vina, or GNINA. For structure-based ligand generation, explore PocketFlow.
DynamicBind uses a diffusion-based generative model that learns to transition proteins from their apo state to ligand-specific holo conformations. The model is E(3)-equivariant, meaning it respects the 3D rotational and translational symmetries of molecular structures. This mathematical property allows the model to train on 1000× less data while achieving superior generalization compared to non-equivariant architectures.
The diffusion process operates over 20 inference steps with progressively smaller perturbations. Rather than adding random Gaussian noise, DynamicBind learns biologically relevant conformational transitions by training on paired apo-holo crystal structures from the PDBbind2020 dataset (19,443 protein-ligand complexes).
Each protein residue is represented as a node with two geometric features: backbone coordinates and side-chain dihedral angles ( angles). This coarse-graining reduces degrees of freedom while maintaining the ability to reconstruct all non-hydrogen atom positions. The model simultaneously predicts:
The use of tensor products of irreducible representations enables the model to capture complex protein-ligand interactions while preserving SE(3) equivariance throughout the neural network layers.
DynamicBind accommodates large protein conformational changes, including kinase DFG-in/out transitions and cryptic pocket opening. By learning smooth energy landscapes between equilibrium states, the model can recover holo-structures even when the initial AlphaFold prediction has large pocket RMSD ( Å) relative to the bound state.
The model outputs both the predicted ligand pose and the corresponding protein conformation, allowing users to visualize how binding site residues adjust to accommodate the ligand.
DynamicBind automatically removes water molecules, non-standard residues, and ligands during preprocessing. For structure prediction, use ESMFold, Boltz-2, or Chai-1.
The ligand is automatically processed and converted to the appropriate internal representation for docking.
Controls how many binding poses to generate per protein-ligand pair. More poses increase the chance of sampling near-native conformations but require longer computation time.
We recommend starting with 10 poses for initial exploration and increasing to 20-40 for final predictions or challenging targets where the binding site is unknown.
The number of diffusion denoising steps used during sampling. Higher values may improve pose quality by allowing more gradual refinement but increase computation time proportionally.
The model was trained and validated with 20 steps, which provides a good balance between quality and speed. Values below 15 may produce lower-quality poses, while values above 25 show diminishing returns.
When enabled, DynamicBind predicts ligand-induced conformational changes in both backbone and side-chain positions. When disabled, the model performs rigid docking with the input structure held fixed.
Enable flexibility (default) when working with apo structures or AlphaFold predictions. Disable for holo structures where the binding site conformation is already optimized, or when you want to test if a ligand can bind without requiring protein adjustments.
DynamicBind returns ranked binding poses with two scoring metrics:
Predicted binding affinity in units, where . Higher values indicate stronger binding:
The affinity prediction is learned from PDBbind experimental binding data and provides an estimate of binding strength. Note that affinity predictions are less reliable for proteins with low sequence homology to the training set.
Confidence metric ranging from 0 to 1 that estimates the accuracy of the predicted protein conformation. Higher lDDT scores indicate the model has high confidence in both the ligand pose and the induced protein conformational change.
Poses are ranked by a combination of lDDT and clash scores to prioritize geometrically plausible structures with favorable predicted confidence.
The top-ranked pose represents the model's best prediction, but examining multiple poses is recommended. For successful predictions on benchmark datasets:
Download predicted structures in SDF format (ligand) and PDB format (protein conformation) for further analysis or molecular dynamics simulations.
DynamicBind is particularly valuable when:
For these applications, DynamicBind achieves 1.7× higher success rates compared to DiffDock under stringent accuracy criteria.
Performance depends on similarity to the PDBbind2020 training set. Predictions for proteins with low sequence homology to known structures show reduced accuracy. The model performs best on major drug target families (kinases, GPCRs, nuclear receptors, ion channels) that are well-represented in crystallographic databases.
When the binding site is unknown, DynamicBind may struggle to identify the correct pocket, especially for novel protein targets. Providing a general binding region or using multiple inference runs with different random seeds can improve coverage.
Perfect pose selection using confidence scores could improve success rates from 33% to 50%, indicating that the current lDDT-based ranking occasionally prioritizes incorrect poses. Manual inspection of top-ranked poses is recommended for critical applications.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.