Related tools
CANYA
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
Prot2Prop
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.
Protein stability
Predict protein stability using validated BioPython methods: Instability Index, Aliphatic Index, GRAVY, flexibility analysis, and charge distribution
ThermoMPNN
Predict protein thermostability changes (ΔΔG) for point mutations using a graph neural network. Enables computational saturation mutagenesis screening to identify stabilizing mutations.
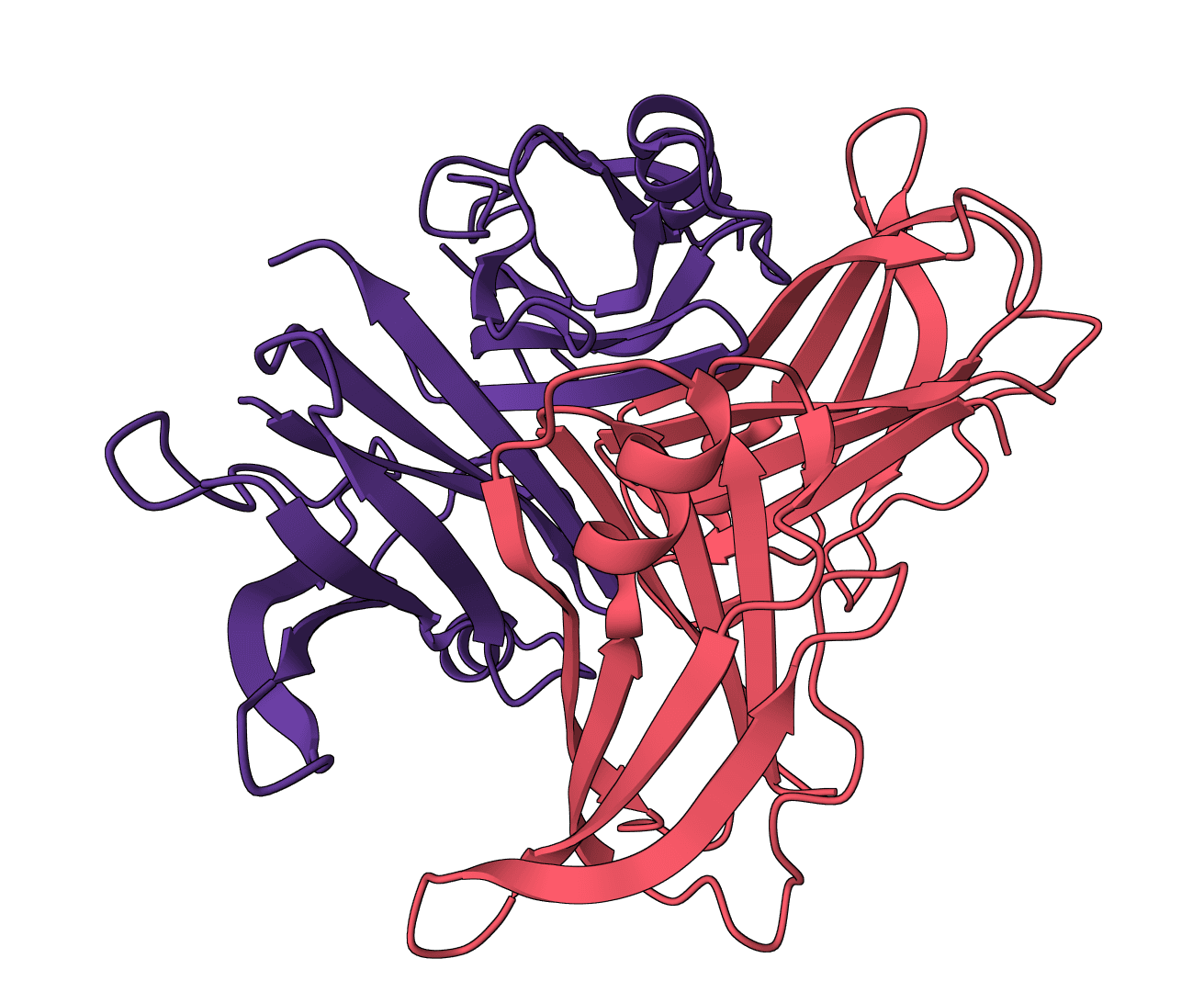
Aggrescan3D
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
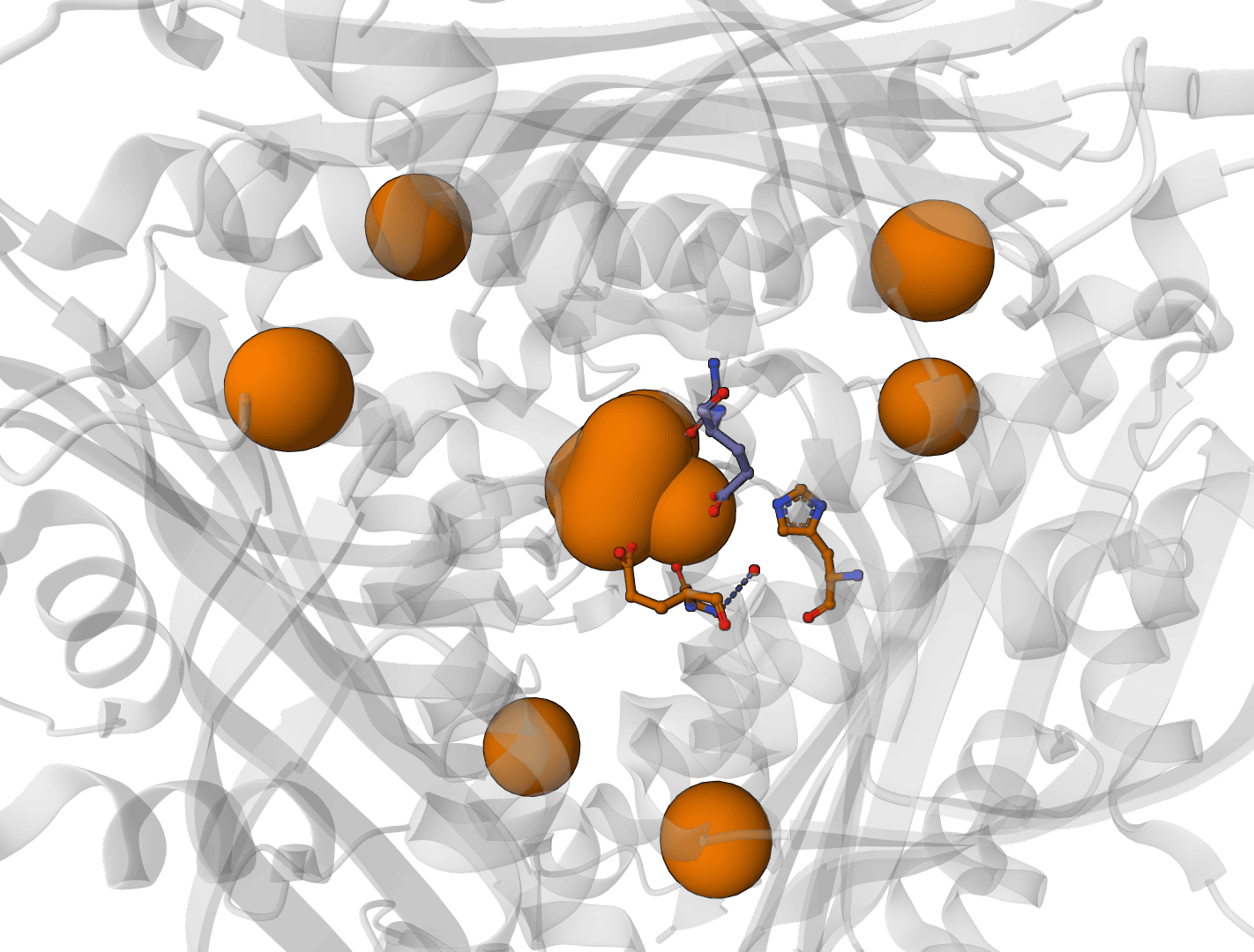
AllMetal3D
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
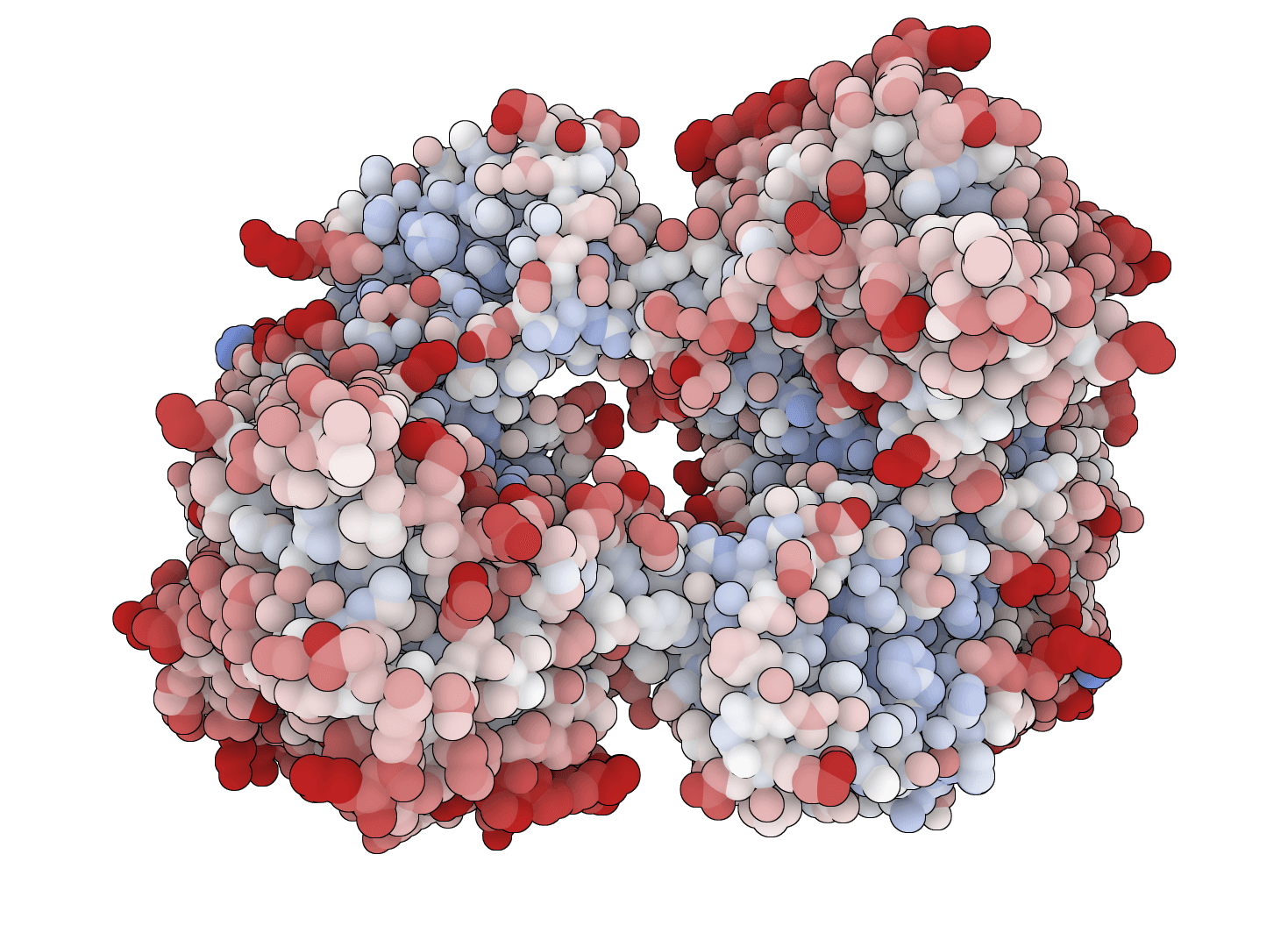
PROPKA 3
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
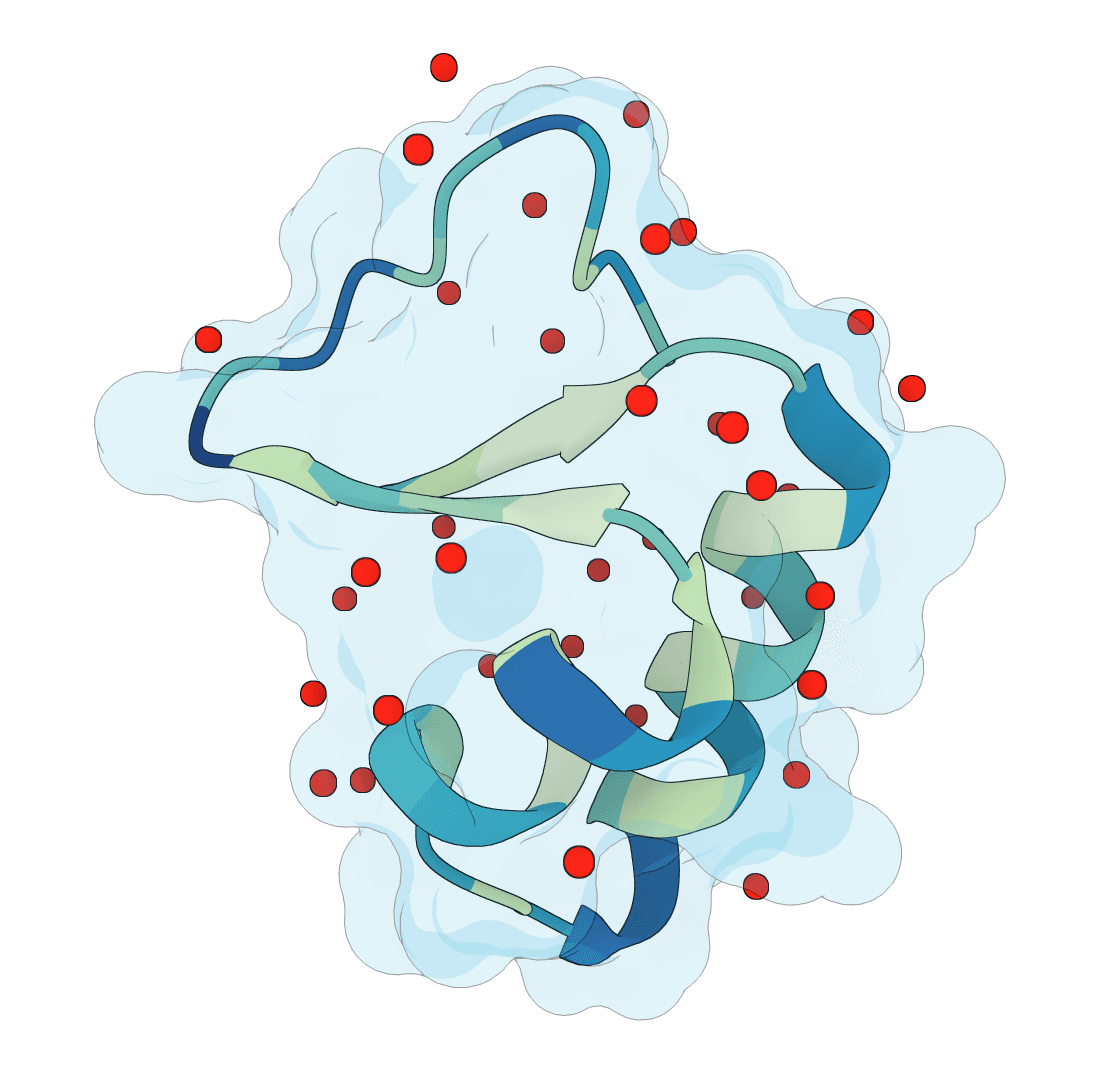
SuperWater
Predict protein hydration sites from a structure using a diffusion model with ESM features and a confidence-filtering head.
AbLang
Restore missing residues in antibody sequences using a language model trained on the Observed Antibody Space (OAS) database. Achieves better restoration than IMGT germlines or ESM-1b while being 7x faster.
IPC 2.0 (isoelectric point calculator)
Isoelectric Point Calculator 2.0 - Predict protein/peptide isoelectric point (pI) using 18+ validated pKa scales, SVR models, and deep learning. Supports proteins, peptides, and comprehensive analysis.