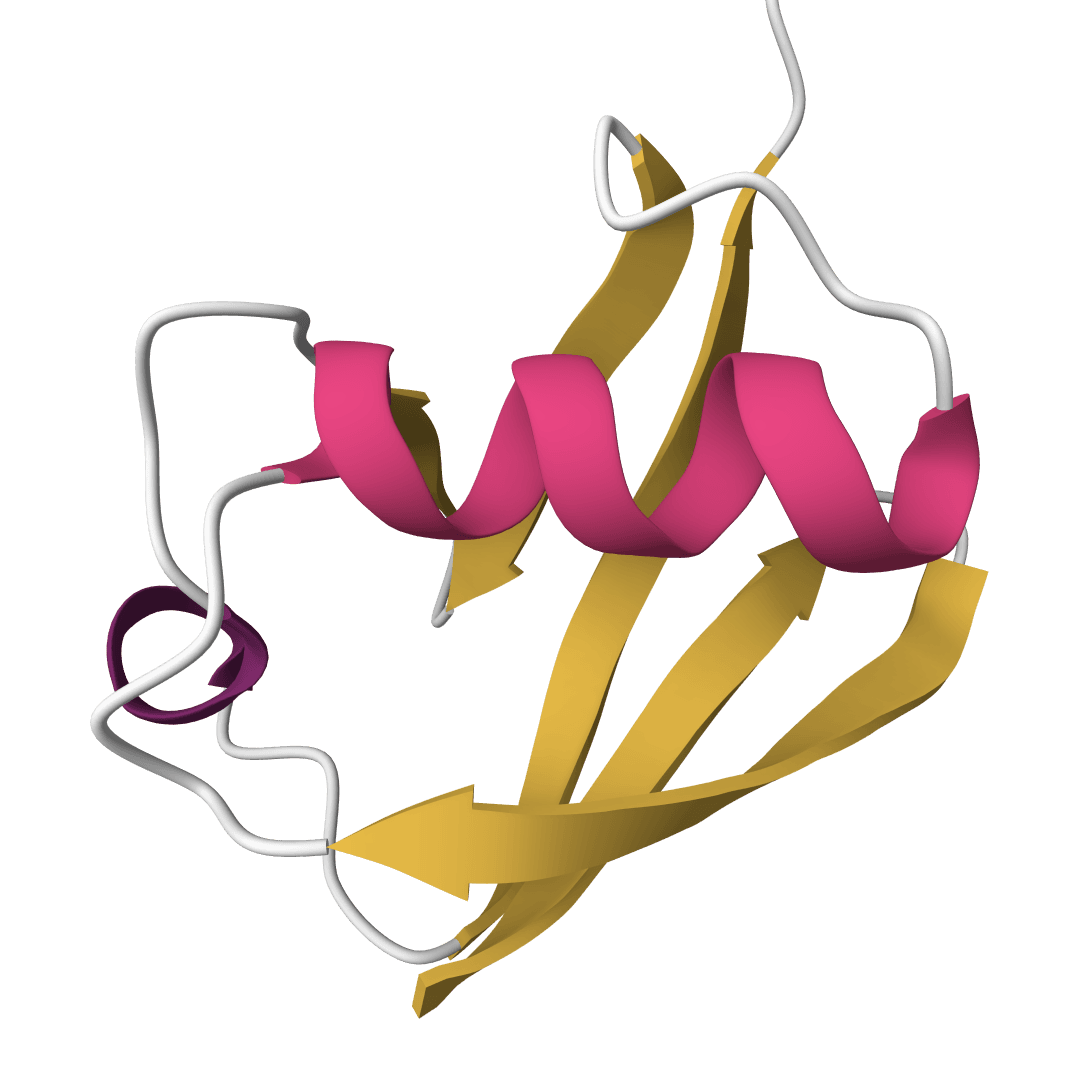
Predict protein secondary structure from amino acid propensities for helices, sheets, and turns. Learn more
Predict protein secondary structure from amino acid propensities for helices, sheets, and turns. Learn more
Predict protein secondary structure from amino acid propensities for helices, sheets, and turns. Learn more
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.

Plot net charge vs pH for protein sequences. Visualize how protein charge changes across pH 0-14 and identify the isoelectric point (pI) where the net charge crosses zero.
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Match experimental peptide masses against theoretical digest fragments of a protein sequence. Identify peptides from mass spectrometry data by peptide mass fingerprinting.
Generate Kyte-Doolittle hydropathy plots to visualize hydrophobic and hydrophilic regions along protein sequences. Identify transmembrane domains and surface-exposed regions.
Generate hydrophobicity plots using 24 different amino acid scales. Visualize hydrophobic and hydrophilic regions for protein analysis, epitope prediction, and membrane protein studies.
Predict protease and chemical cleavage sites across a protein sequence for up to 39 enzymes simultaneously. Identify where each enzyme cuts, the cleavage residue, and context window around each site.
Cleave a protein sequence with a chosen protease and compute the masses of the resulting peptides. Supports multiple enzymes, missed cleavages, chemical modifications, and different ion types for mass spectrometry experiment planning.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Plot net charge vs pH for protein sequences. Visualize how protein charge changes across pH 0-14 and identify the isoelectric point (pI) where the net charge crosses zero.
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Match experimental peptide masses against theoretical digest fragments of a protein sequence. Identify peptides from mass spectrometry data by peptide mass fingerprinting.
Generate Kyte-Doolittle hydropathy plots to visualize hydrophobic and hydrophilic regions along protein sequences. Identify transmembrane domains and surface-exposed regions.
Generate hydrophobicity plots using 24 different amino acid scales. Visualize hydrophobic and hydrophilic regions for protein analysis, epitope prediction, and membrane protein studies.
Predict protease and chemical cleavage sites across a protein sequence for up to 39 enzymes simultaneously. Identify where each enzyme cuts, the cleavage residue, and context window around each site.
Cleave a protein sequence with a chosen protease and compute the masses of the resulting peptides. Supports multiple enzymes, missed cleavages, chemical modifications, and different ion types for mass spectrometry experiment planning.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
Predict multiple protein developability properties from amino-acid sequences using a multitask ProstT5 adapter.
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Insulin B-chain
The Chou-Fasman method predicts protein secondary structure from amino acid sequence alone. Published by Peter Chou and Gerald Fasman in 1974, it was the first widely used algorithm to show that local amino acid composition carries enough information to predict helices, sheets, and turns.
The approach is statistical rather than physical. Chou and Fasman counted how often each amino acid appeared in known crystal structures within helices, sheets, or turns, then converted those frequencies into propensity scores. A propensity above 100 means that amino acid favors the structure; below 100 means it disfavors it. These 60 numbers (20 amino acids times 3 structure types) are the entire model.
Accuracy sits around 60-65%, well below modern deep learning methods like ProstT5 or ESMFold. The method remains useful as a teaching tool and as a fast, interpretable baseline. Because it uses no MSA, no neural network, and no GPU, predictions return in seconds.
ProteinIQ runs the canonical Chou-Fasman algorithm (1978 parameter set) on its servers. Paste one or more sequences and results are returned within seconds.
| Input | Description |
|---|---|
Protein sequence | One or more sequences in FASTA format, raw text, or uploaded as .fasta/.txt/.csv/.pdb files. |
Batch fetch | Fetch sequences by PDB ID from RCSB. |
| Column | Description |
|---|---|
Structure | Full per-residue prediction string using H (helix), E (sheet), T (turn), C (coil). |
Helix % / Sheet % / Turn % / Coil % | Fraction of residues assigned to each state. |
Helix # / Sheet # / Turn # | Number of distinct regions of each type. |
Helix avg / Sheet avg / Turn avg | Mean length (in residues) of each region type. |
Results can be downloaded as CSV, JSON, or copied directly.
The algorithm runs in four steps, applied sequentially to each sequence.
The method scans for local stretches rich in structure-forming residues:
These thresholds reflect the cooperative nature of secondary structure formation. A single strong former surrounded by breakers won't nucleate.
Each nucleated region extends outward one residue at a time in both directions. Extension continues as long as the average propensity of 4 consecutive residues at the boundary stays above 100. Once this running average drops below the threshold, the region terminates.
Turns are predicted for each tetrapeptide (4-residue window) that satisfies all three conditions:
When predicted helix and sheet regions overlap, the algorithm compares the total helix propensity () against total sheet propensity () across the overlap. The higher sum wins. After resolution, any helix or sheet region shorter than 5 residues is removed as unreliable.
Positions not assigned to helix, sheet, or turn default to random coil (C).
The Structure string is the primary output. Each character maps directly to a position in the input sequence:
Typical globular proteins show 30-40% helix and 15-25% sheet. If a prediction shows 0% for both, the sequence may be intrinsically disordered, or too short for meaningful nucleation (sequences under ~20 residues often produce all-coil predictions).
The percentage and region count columns are useful for comparing across sequences. A protein with 35% helix in 4 regions has a different architecture than one with 35% helix in 1 long region.
Chou-Fasman is a sequence-only, statistics-only method. Several alternatives exist depending on what matters most:
Chou-Fasman's niche is speed and interpretability. Every prediction can be traced back to a specific propensity value and no black-box model is involved. This makes it well-suited for teaching the fundamentals of structure prediction, for quickly screening large sequence sets, and for situations where understanding why a prediction was made matters more than accuracy.