Perform maximum-likelihood phylogenetic tree inference with RAxML-NG for aligned protein or DNA sequences. Supports ML search, bootstrap analysis, and native automatic model-family selection.
DR-BERT is a compact protein language model that predicts intrinsically disordered regions (IDRs) in proteins. It outputs per-residue disorder probability scores (0–1) from amino acid sequences, enabling fast and accurate annotation of disordered regions without structural data.
Restore missing antibody residues, generate 768-dimensional sequence or residue representations, and calculate amino-acid likelihood scores with the original AbLang heavy- and light-chain models.
Antibody-specific language model for predicting non-germline residues (NGL) in antibody sequences. AbLang-2 addresses germline bias in existing antibody language models by focusing on somatic hypermutation patterns, enabling more accurate prediction of amino acid likelihoods and generation of context-aware embeddings for antibody sequences.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
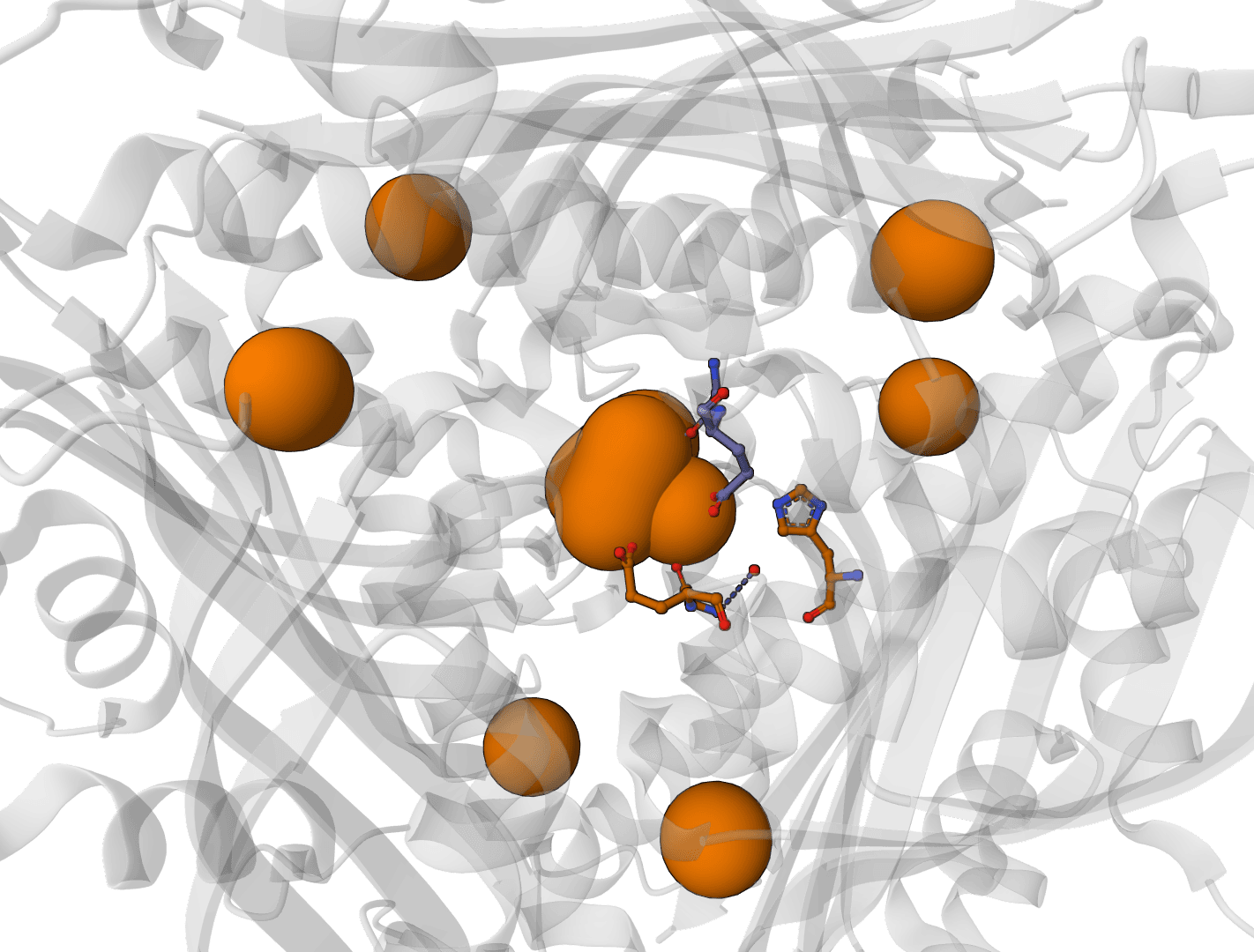
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
DeepEMhancer is a deep learning-based post-processing tool for cryo-EM maps. It performs automatic sharpening, masking, and denoising in a single step without requiring an atomic model. Supports half-map inputs for improved local mask estimation.
ESM-2 is a 650M parameter protein language model from Meta AI trained on 250M protein sequences. Generate rich sequence representations for downstream tasks like structure prediction, function annotation, and variant effect prediction.
ESM-C generates protein sequence representations and optional forward-pass sequence logits using Biohub protein language models. It supports the 300M, 600M, and 6B model variants for embedding extraction from tokenizer-compatible protein sequences.
Perform maximum-likelihood phylogenetic tree inference with RAxML-NG for aligned protein or DNA sequences. Supports ML search, bootstrap analysis, and native automatic model-family selection.
DR-BERT is a compact protein language model that predicts intrinsically disordered regions (IDRs) in proteins. It outputs per-residue disorder probability scores (0–1) from amino acid sequences, enabling fast and accurate annotation of disordered regions without structural data.
Restore missing antibody residues, generate 768-dimensional sequence or residue representations, and calculate amino-acid likelihood scores with the original AbLang heavy- and light-chain models.
Antibody-specific language model for predicting non-germline residues (NGL) in antibody sequences. AbLang-2 addresses germline bias in existing antibody language models by focusing on somatic hypermutation patterns, enabling more accurate prediction of amino acid likelihoods and generation of context-aware embeddings for antibody sequences.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
Predict protein aggregation nucleation propensity from amino acid sequences using the Lehner Lab CANYA neural network.
DeepEMhancer is a deep learning-based post-processing tool for cryo-EM maps. It performs automatic sharpening, masking, and denoising in a single step without requiring an atomic model. Supports half-map inputs for improved local mask estimation.
ESM-2 is a 650M parameter protein language model from Meta AI trained on 250M protein sequences. Generate rich sequence representations for downstream tasks like structure prediction, function annotation, and variant effect prediction.
ESM-C generates protein sequence representations and optional forward-pass sequence logits using Biohub protein language models. It supports the 300M, 600M, and 6B model variants for embedding extraction from tokenizer-compatible protein sequences.
Configure inputs to begin
Set options on the left, then click “Run Clustering”.
AF-Cluster is a method for predicting multiple protein conformations by clustering a multiple sequence alignment (MSA) before running AlphaFold2. Standard AlphaFold2 predictions converge on a single dominant structure, even for proteins that adopt two or more biologically relevant folds. AF-Cluster addresses this by splitting the MSA into sequence subgroups using the DBSCAN density-based clustering algorithm, then generating separate AlphaFold2 predictions from each cluster.
The approach was developed by Hannah Wayment-Steele and colleagues and published in Nature (volume 625) in 2024. The authors validated the method on metamorphic proteins, including the cyanobacterial clock protein KaiB, where AF-Cluster correctly predicted both the ground-state and fold-switched conformations. NMR spectroscopy confirmed that a KaiB variant predicted by AF-Cluster was indeed stabilized in the opposite fold.
Proteins evolve under selective pressure to maintain function, and function often requires switching between conformational states. Homologous sequences in an MSA may carry co-evolutionary signals for different conformations. When the full MSA is fed to AlphaFold2, these conflicting signals average out and the prediction collapses onto a single state.
AF-Cluster separates these signals by clustering the MSA:
- characters is strictly below the selected cutoff.min_samples sequences; sequences outside dense regions are labeled as noise.Each cluster's alignment can then be used as input to AlphaFold2 independently, producing structure predictions that may capture different conformational states.
ProteinIQ runs the commit-pinned AF-Cluster source on cloud infrastructure, so no software installation is needed. The current source revision is 6b22451. The runtime applies the identified proteiniq-cli-numeric-types-v1 parser correction and verifies the original and corrected entrypoint digests before publishing the image.
| Input | Description |
|---|---|
Multiple Sequence Alignment | A protein MSA in FASTA or A3M format. The first sequence is the query. After lowercase A3M insertions are removed, all records must have the same aligned-column count and use the 20 standard amino acids plus gaps. At least 10 non-query sequences must remain after gap filtering. Jobs accept up to 5,000 sequences, 2,000,000 aligned sequence cells, and 5 MB of submitted text. |
MSAs can be generated from tools like Clustal Omega or MAFFT, or obtained from databases such as UniRef or ColabFold search.
| Setting | Description |
|---|---|
Min samples per cluster | Minimum number of sequences required to form a DBSCAN cluster (2–20, default 3). Higher values produce fewer, more populated clusters. |
Gap fraction cutoff | Remove each non-query sequence when its aligned-column gap fraction is at or above this value (0.05–1, default 0.25). Lower values enforce stricter filtering. |
DBSCAN epsilon | Maximum Euclidean distance in one-hot encoded alignment space for DBSCAN neighborhood membership (0–20, default 0 for automatic). At 0, AF-Cluster scans 3–20 in 0.5 increments. |
Minimum epsilon | First epsilon tested during automatic scanning (0.1–50, default 3). |
Maximum epsilon | Largest epsilon available to automatic scanning (0.1–50, default 20). It must be greater than or equal to the minimum. |
Epsilon step | Increment between automatic epsilon candidates (0.1–5, default 0.5). Automatic scans are limited to 100 candidates to keep runtime bounded. |
| Setting | Description |
|---|---|
Generate PCA plot | Project clustered sequences onto their first two principal components. Useful for seeing how clusters separate in sequence space. |
AF-Cluster produces several files:
_REF.a3m file described in its README.All of these artifacts are available to workflows. Cluster and control alignments are emitted as separate sequence artifacts so each alignment can be connected directly to a downstream folding step.
The on-screen assignments table previews up to 1,000 rows. The downloadable clustering-assignments TSV always retains every source row.
AF-Cluster is most valuable for proteins suspected of adopting multiple folds: