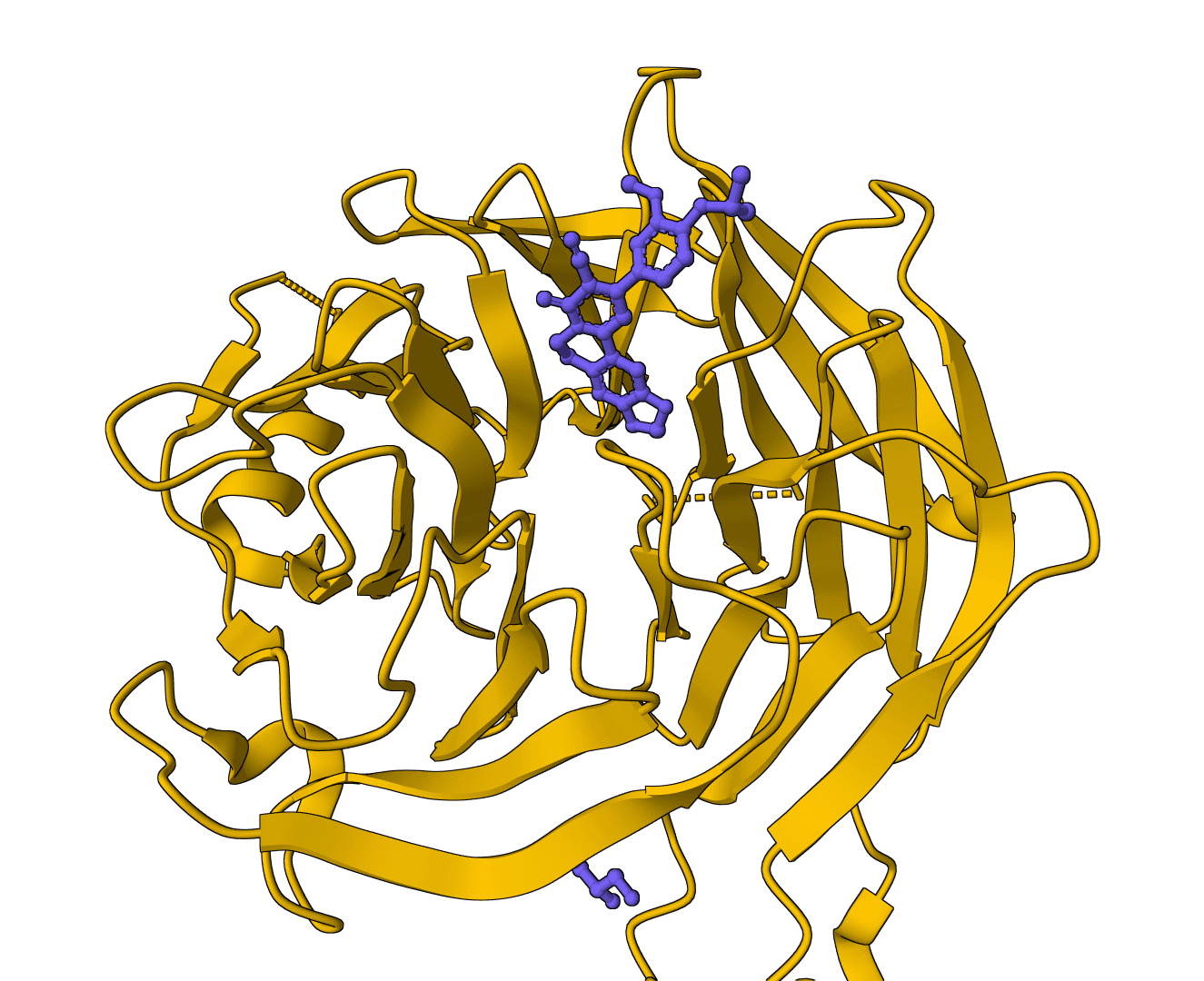
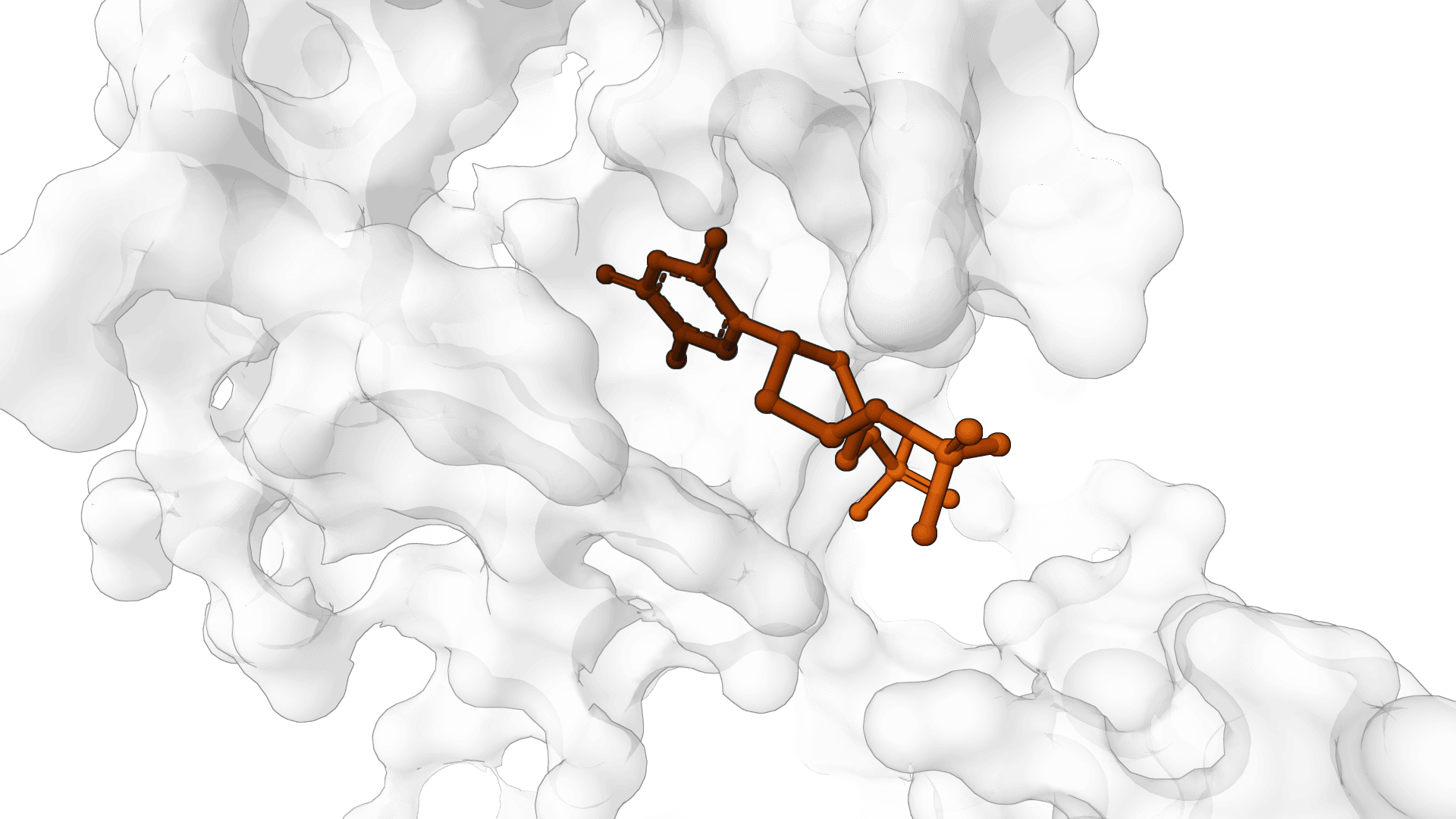
fpocket is an open-source protein pocket detection algorithm that identifies ligand binding sites using Voronoi tessellation and alpha sphere geometry.
The software analyzes protein structures to locate and characterize cavities and clefts where small molecules can bind. The algorithm ranks detected pockets by druggability and provides geometric and physicochemical descriptors.
Traditional binding site detection methods often rely on grid-based approaches or require ligand-bound structures as templates. fpocket instead uses geometric principles to identify pockets directly from protein coordinates, making it applicable to both bound and unbound (apo) protein structures.
The algorithm achieves 94% detection accuracy within the top three ranked pockets while executing in under 3 seconds per structure.
ProteinIQ provides a web interface for running fpocket without command-line installation. You upload a protein structure or enter a PDB ID, and the tool returns ranked pockets with geometric and chemical descriptors.
| Input | Description |
|---|---|
Protein Structure | The target protein for pocket detection. Upload a PDB or mmCIF file, or enter a 4-character PDB ID (e.g., 1HSG) to fetch from RCSB. Maximum file size is 50 MB. |
The default settings run automatic fpocket pocket detection. Optional controls can focus the analysis on known sites, selected chains, or adjusted alpha-sphere clustering parameters.
| Setting | Default | Description |
|---|---|---|
Site mode | Automatic pocket detection | Use automatic detection, a known ligand residue, a residue-defined pocket, or a chain treated as the ligand. |
Ligand residue | Empty | Known ligand format for focused scoring, such as 1224:PU8:A. |
Pocket residues | Empty | Dot-separated residue tokens for an explicit pocket, such as 107::A.138::A.51::A. |
Model number | 0 | Use all models, or select a specific model number. |
Chain filter | All chains | Keep or drop compact chain lists such as A,C or B,D. |
Structure output | fpocket default | Choose fpocket's default structure output mode, PDB only, mmCIF only, or both. |
Alpha sphere radius | 3.4 to 6.2 | Minimum and maximum alpha sphere radii used during candidate sphere filtering. |
Minimum spheres per pocket | 15 | Minimum alpha spheres required for a detected pocket. |
Minimum apolar sphere ratio | 0 | Minimum apolar alpha sphere ratio for retained pockets. |
Clustering distance | 2.4 | Distance threshold used when clustering alpha spheres into pockets. |
Clustering method | Single linkage | Hierarchical clustering method for pocket grouping. |
Distance measure | Euclidean | Distance metric used by clustering. |
Apolar alpha spheres threshold | 3 | Threshold for apolar alpha sphere count during pocket detection. |
Monte Carlo volume iterations | 300 | Iterations used for volume estimation. |
fpocket returns a ranked table of detected pockets with the standard descriptors written to the *_info.txt report.
| Column | Description |
|---|---|
Pocket | Pocket rank, where 1 is the top-ranked predicted site. |
Score | Overall fpocket pocket score derived from geometric and physicochemical descriptors. |
Drug Score | Druggability score estimating suitability for small-molecule binding. |
Volume (A^3) | Pocket volume in cubic angstroms. |
Apolar SASA (A^2) | Solvent-accessible surface area assigned to apolar pocket surface. |
Polar SASA (A^2) | Solvent-accessible surface area assigned to polar pocket surface. |
Alpha Spheres | Number of alpha spheres assigned to the pocket. |
Total SASA (A^2) | Total solvent-accessible surface area of the pocket. |
Mean Local Hydrophobic Density | Average local hydrophobic density around pocket alpha spheres. |
Mean Alpha Sphere Radius | Mean radius of alpha spheres assigned to the pocket. |
Mean Alpha Sphere Solvent Access | Average solvent accessibility of pocket alpha spheres. |
Apolar Alpha Sphere Proportion | Fraction of alpha spheres classified as apolar. |
Hydrophobicity Score | Aggregate pocket hydrophobicity descriptor reported by fpocket. |
Volume Score | fpocket's internal volume-related score term. |
Polarity Score | Integer polarity descriptor based on lining residue properties. |
Charge Score | Integer net charge descriptor at pH 7 from lining residues. |
Proportion of Polar Atoms | Fraction of pocket atoms considered polar. |
Alpha Sphere Density | Pocket density descriptor based on alpha sphere spacing. |
Centroid-Alpha Sphere Max Dist | Distance from the pocket center of mass to the farthest alpha sphere. |
Flexibility | fpocket flexibility descriptor derived from B-factors. |
The Files tab includes the fpocket-generated artifacts for the run, including the *_info.txt report, combined pocket structures, per-pocket coordinate files, visualization scripts, and a CSV export of the spreadsheet values.
The druggability score integrates multiple descriptors using a partial least squares model trained on known drug-binding sites:
Scores reflect both geometric properties (pocket depth, volume) and chemical features (hydrophobicity, polarity balance).
fpocket employs alpha sphere theory based on computational geometry. An alpha sphere is defined as a sphere that touches exactly four protein atoms on its boundary while containing no atoms in its interior. These spheres naturally concentrate in protein cavities and clefts, making them ideal markers for binding site detection.
The algorithm begins by computing a Voronoi decomposition of 3D space around the protein using the Qhull library. Voronoi vertices—points equidistant from four neighboring atoms—correspond to potential alpha sphere centers. fpocket filters these vertices by radius, discarding spheres too small (tight atomic packing in the protein core) or too large (solvent-exposed surface regions).
The default radius range is 3.4–6.2 Å, optimized for typical small molecule binding sites. This geometric criterion eliminates ~80% of candidate spheres before clustering.
fpocket groups neighboring alpha spheres into pockets using a three-pass clustering procedure:
This hierarchical approach handles irregular pocket geometries better than single-threshold methods. The algorithm leverages Qhull's neighbor lists to avoid pairwise distance calculations, achieving near-linear runtime scaling.
Each pocket receives a composite score derived from five weighted descriptors:
| Descriptor | Weight | Meaning |
|---|---|---|
| Normalized alpha sphere count | 0.3 | Pocket size indicator |
| Mean local hydrophobic density | 0.25 | Apolar residue concentration |
| Proportion of apolar spheres | 0.2 | Hydrophobic character |
| Polarity score | 0.15 | Polar residue presence |
| Alpha sphere density | 0.1 | Spatial compactness |
Weights derive from partial least squares regression against a training set of 48 known ligand-binding sites. The scoring function prioritizes deep, hydrophobic pockets with balanced polarity—characteristics of successful drug targets.
Beyond the scoring function, fpocket computes additional physicochemical properties:
These descriptors enable users to assess pockets beyond the druggability score alone.
Benchmark studies on the PocketPicker dataset (48 diverse proteins) demonstrate:
On the Astex Diverse set (85 high-quality pharmaceutical complexes):
The algorithm outperforms CAST, PASS, SURFNET, and LIGSITE at rank-3 while executing 10–100× faster than grid-based competitors. This speed advantage enables proteome-scale screening applications.
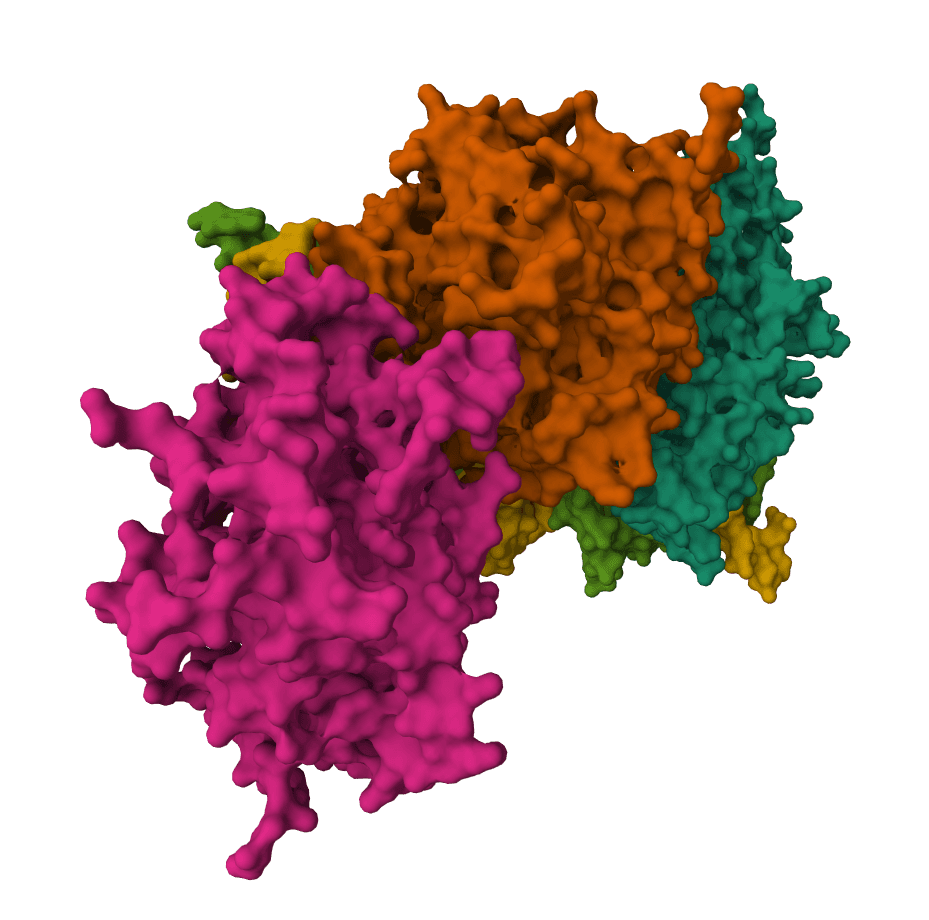
AF2BIND predicts ligand-binding residues from a protein structure using AlphaFold2 pair representations and a 20-residue bait sequence.
Rank a compound library against one protein target with SPRINT protein and ligand co-embeddings and native cosine similarity.
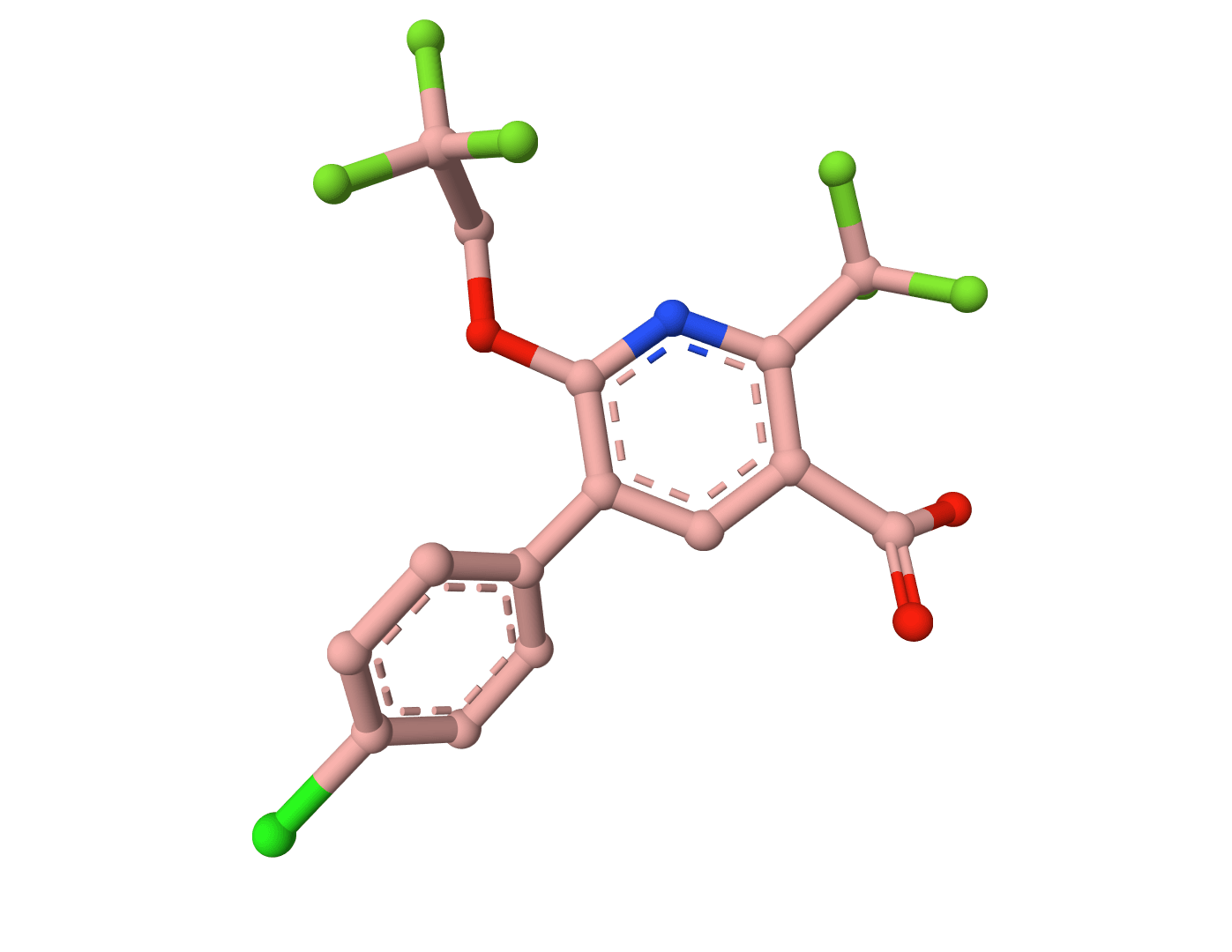
Lipinski's Rule of Five predicts whether compounds will be orally bioavailable by evaluating molecular weight, LogP, hydrogen bond donors, and acceptors.
Predict protein solubility and usability for E. coli expression using ESM protein language models
Predict ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties from SMILES strings using machine learning models trained on Therapeutics Data Commons datasets.
Predict 22 ADMET properties from SMILES strings with the native Admetica Chemprop models from Datagrok.
Identify toxic, reactive, and pharmacokinetically problematic molecular fragments using structural alert patterns
Predict toxicity and synthetic accessibility of small molecules using machine learning. eToxPred combines toxicity risk assessment with synthetic accessibility scoring to help prioritize drug candidates.
Screen for lead-like compounds using stricter molecular descriptor criteria than Lipinski or Veber rules for early-stage drug discovery
Screen compounds for Pan-Assay Interference patterns that cause false positives in biological assays
fpocket is an open-source protein pocket detection algorithm that identifies ligand binding sites using Voronoi tessellation and alpha sphere geometry.
The software analyzes protein structures to locate and characterize cavities and clefts where small molecules can bind. The algorithm ranks detected pockets by druggability and provides geometric and physicochemical descriptors.
Traditional binding site detection methods often rely on grid-based approaches or require ligand-bound structures as templates. fpocket instead uses geometric principles to identify pockets directly from protein coordinates, making it applicable to both bound and unbound (apo) protein structures.
The algorithm achieves 94% detection accuracy within the top three ranked pockets while executing in under 3 seconds per structure.
ProteinIQ provides a web interface for running fpocket without command-line installation. You upload a protein structure or enter a PDB ID, and the tool returns ranked pockets with geometric and chemical descriptors.
| Input | Description |
|---|---|
Protein Structure | The target protein for pocket detection. Upload a PDB or mmCIF file, or enter a 4-character PDB ID (e.g., 1HSG) to fetch from RCSB. Maximum file size is 50 MB. |
The default settings run automatic fpocket pocket detection. Optional controls can focus the analysis on known sites, selected chains, or adjusted alpha-sphere clustering parameters.
| Setting | Default | Description |
|---|---|---|
Site mode | Automatic pocket detection | Use automatic detection, a known ligand residue, a residue-defined pocket, or a chain treated as the ligand. |
Ligand residue | Empty | Known ligand format for focused scoring, such as 1224:PU8:A. |
Pocket residues | Empty | Dot-separated residue tokens for an explicit pocket, such as 107::A.138::A.51::A. |
Model number | 0 | Use all models, or select a specific model number. |
Chain filter | All chains | Keep or drop compact chain lists such as A,C or B,D. |
Structure output | fpocket default | Choose fpocket's default structure output mode, PDB only, mmCIF only, or both. |
Alpha sphere radius | 3.4 to 6.2 | Minimum and maximum alpha sphere radii used during candidate sphere filtering. |
Minimum spheres per pocket | 15 | Minimum alpha spheres required for a detected pocket. |
Minimum apolar sphere ratio | 0 | Minimum apolar alpha sphere ratio for retained pockets. |
Clustering distance | 2.4 | Distance threshold used when clustering alpha spheres into pockets. |
Clustering method | Single linkage | Hierarchical clustering method for pocket grouping. |
Distance measure | Euclidean | Distance metric used by clustering. |
Apolar alpha spheres threshold | 3 | Threshold for apolar alpha sphere count during pocket detection. |
Monte Carlo volume iterations | 300 | Iterations used for volume estimation. |
fpocket returns a ranked table of detected pockets with the standard descriptors written to the *_info.txt report.
| Column | Description |
|---|---|
Pocket | Pocket rank, where 1 is the top-ranked predicted site. |
Score | Overall fpocket pocket score derived from geometric and physicochemical descriptors. |
Drug Score | Druggability score estimating suitability for small-molecule binding. |
Volume (A^3) | Pocket volume in cubic angstroms. |
Apolar SASA (A^2) | Solvent-accessible surface area assigned to apolar pocket surface. |
Polar SASA (A^2) | Solvent-accessible surface area assigned to polar pocket surface. |
Alpha Spheres | Number of alpha spheres assigned to the pocket. |
Total SASA (A^2) | Total solvent-accessible surface area of the pocket. |
Mean Local Hydrophobic Density | Average local hydrophobic density around pocket alpha spheres. |
Mean Alpha Sphere Radius | Mean radius of alpha spheres assigned to the pocket. |
Mean Alpha Sphere Solvent Access | Average solvent accessibility of pocket alpha spheres. |
Apolar Alpha Sphere Proportion | Fraction of alpha spheres classified as apolar. |
Hydrophobicity Score | Aggregate pocket hydrophobicity descriptor reported by fpocket. |
Volume Score | fpocket's internal volume-related score term. |
Polarity Score | Integer polarity descriptor based on lining residue properties. |
Charge Score | Integer net charge descriptor at pH 7 from lining residues. |
Proportion of Polar Atoms | Fraction of pocket atoms considered polar. |
Alpha Sphere Density | Pocket density descriptor based on alpha sphere spacing. |
Centroid-Alpha Sphere Max Dist | Distance from the pocket center of mass to the farthest alpha sphere. |
Flexibility | fpocket flexibility descriptor derived from B-factors. |
The Files tab includes the fpocket-generated artifacts for the run, including the *_info.txt report, combined pocket structures, per-pocket coordinate files, visualization scripts, and a CSV export of the spreadsheet values.
The druggability score integrates multiple descriptors using a partial least squares model trained on known drug-binding sites:
Scores reflect both geometric properties (pocket depth, volume) and chemical features (hydrophobicity, polarity balance).
fpocket employs alpha sphere theory based on computational geometry. An alpha sphere is defined as a sphere that touches exactly four protein atoms on its boundary while containing no atoms in its interior. These spheres naturally concentrate in protein cavities and clefts, making them ideal markers for binding site detection.
The algorithm begins by computing a Voronoi decomposition of 3D space around the protein using the Qhull library. Voronoi vertices—points equidistant from four neighboring atoms—correspond to potential alpha sphere centers. fpocket filters these vertices by radius, discarding spheres too small (tight atomic packing in the protein core) or too large (solvent-exposed surface regions).
The default radius range is 3.4–6.2 Å, optimized for typical small molecule binding sites. This geometric criterion eliminates ~80% of candidate spheres before clustering.
fpocket groups neighboring alpha spheres into pockets using a three-pass clustering procedure:
This hierarchical approach handles irregular pocket geometries better than single-threshold methods. The algorithm leverages Qhull's neighbor lists to avoid pairwise distance calculations, achieving near-linear runtime scaling.
Each pocket receives a composite score derived from five weighted descriptors:
| Descriptor | Weight | Meaning |
|---|---|---|
| Normalized alpha sphere count | 0.3 | Pocket size indicator |
| Mean local hydrophobic density | 0.25 | Apolar residue concentration |
| Proportion of apolar spheres | 0.2 | Hydrophobic character |
| Polarity score | 0.15 | Polar residue presence |
| Alpha sphere density | 0.1 | Spatial compactness |
Weights derive from partial least squares regression against a training set of 48 known ligand-binding sites. The scoring function prioritizes deep, hydrophobic pockets with balanced polarity—characteristics of successful drug targets.
Beyond the scoring function, fpocket computes additional physicochemical properties:
These descriptors enable users to assess pockets beyond the druggability score alone.
Benchmark studies on the PocketPicker dataset (48 diverse proteins) demonstrate:
On the Astex Diverse set (85 high-quality pharmaceutical complexes):
The algorithm outperforms CAST, PASS, SURFNET, and LIGSITE at rank-3 while executing 10–100× faster than grid-based competitors. This speed advantage enables proteome-scale screening applications.
AF2BIND predicts ligand-binding residues from a protein structure using AlphaFold2 pair representations and a 20-residue bait sequence.
Rank a compound library against one protein target with SPRINT protein and ligand co-embeddings and native cosine similarity.
Lipinski's Rule of Five predicts whether compounds will be orally bioavailable by evaluating molecular weight, LogP, hydrogen bond donors, and acceptors.
Predict protein solubility and usability for E. coli expression using ESM protein language models
Predict ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties from SMILES strings using machine learning models trained on Therapeutics Data Commons datasets.
Predict 22 ADMET properties from SMILES strings with the native Admetica Chemprop models from Datagrok.
Identify toxic, reactive, and pharmacokinetically problematic molecular fragments using structural alert patterns
Predict toxicity and synthetic accessibility of small molecules using machine learning. eToxPred combines toxicity risk assessment with synthetic accessibility scoring to help prioritize drug candidates.
Screen for lead-like compounds using stricter molecular descriptor criteria than Lipinski or Veber rules for early-stage drug discovery
Screen compounds for Pan-Assay Interference patterns that cause false positives in biological assays