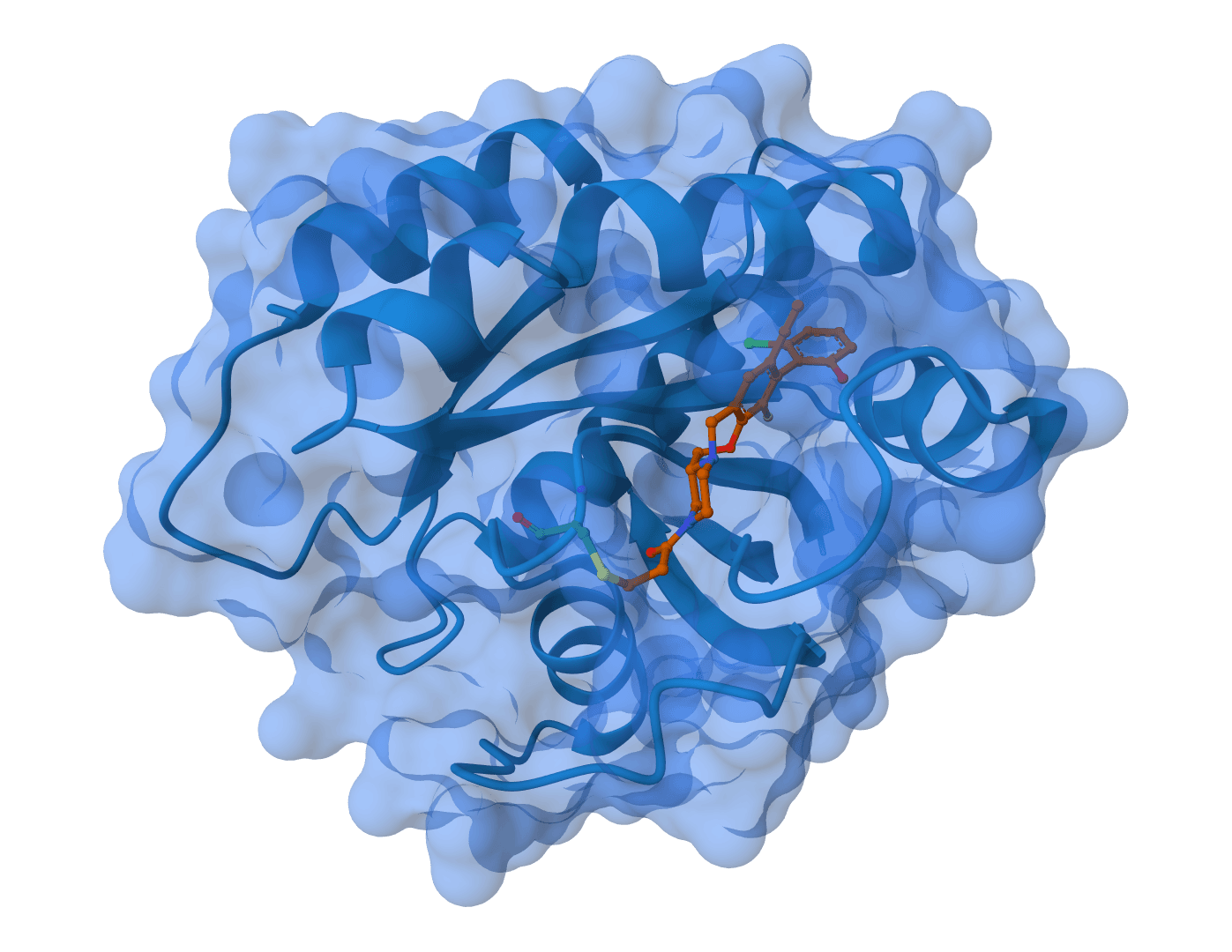
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
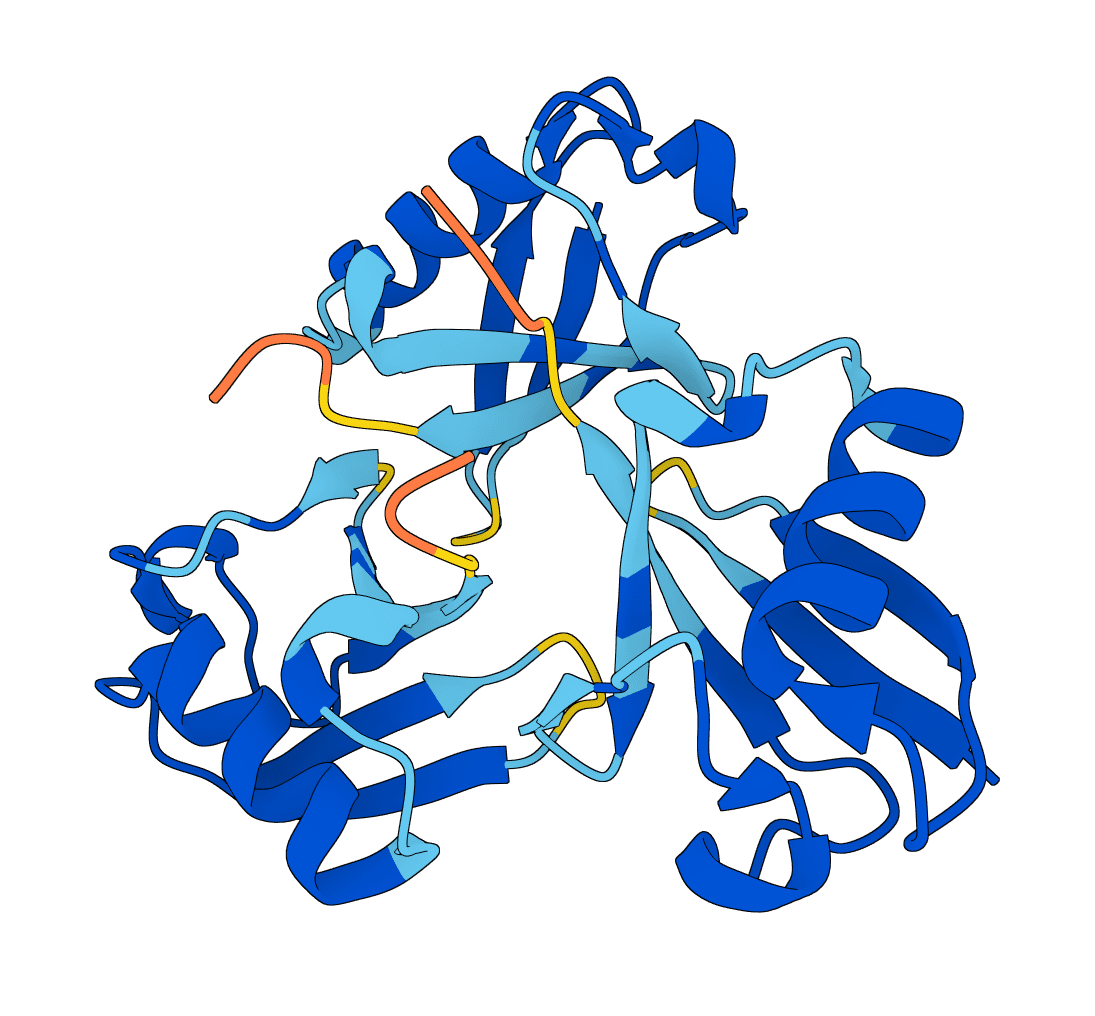
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.

Build all-atom peptide PDB structures from amino acid sequences using PeptideBuilder geometry defaults, with optional backbone angle controls for simple model peptides.
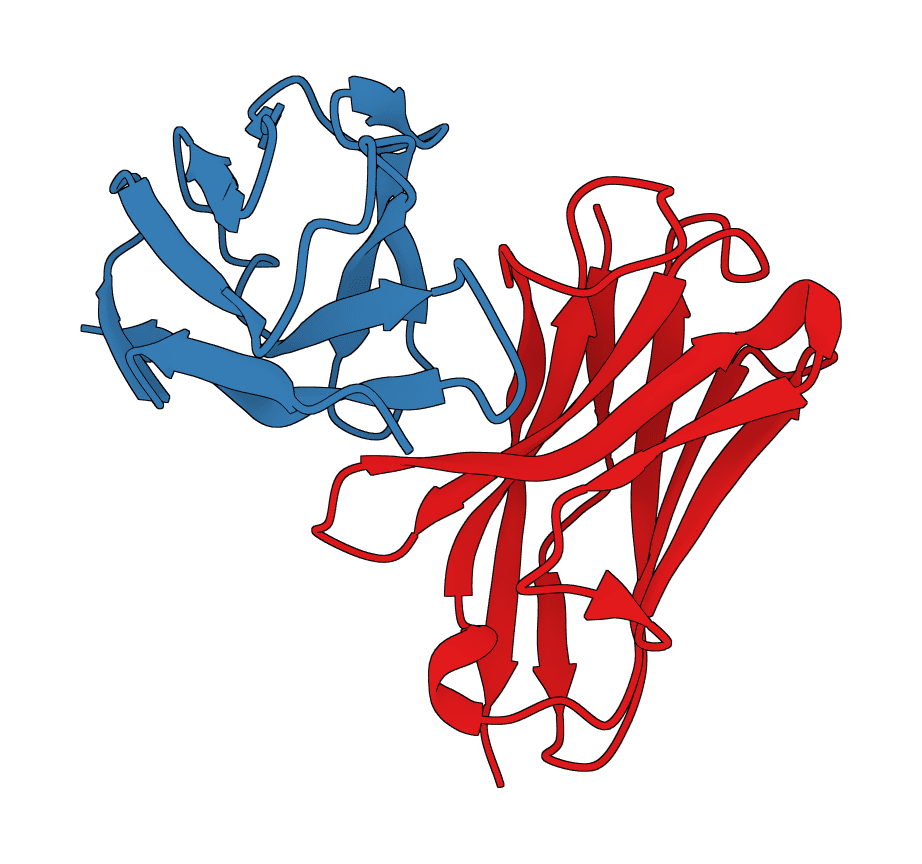
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
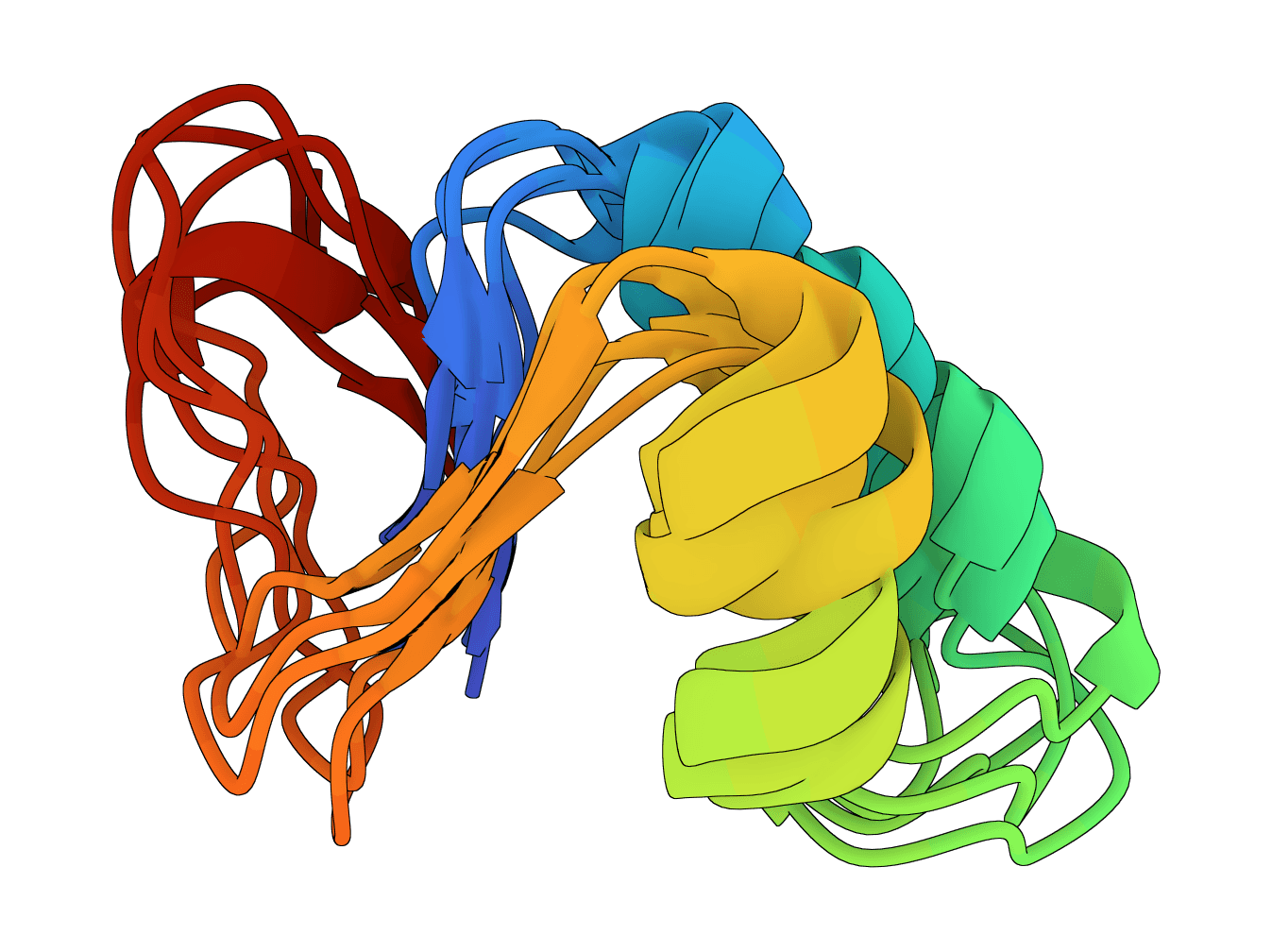
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
Build all-atom peptide PDB structures from amino acid sequences using PeptideBuilder geometry defaults, with optional backbone angle controls for simple model peptides.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Configure inputs to begin
Set options on the left, then click “Submit job”.
HighFold predicts the three-dimensional structures of cyclic peptides — molecules where the chain loops back on itself through a head-to-tail peptide bond, sometimes reinforced by disulfide bridges. Standard protein structure prediction methods like AlphaFold 2 treat sequences as linear chains and encode residue positions accordingly, which means they systematically misrepresent the topology of cyclic peptides.
HighFold solves this by replacing AlphaFold's linear position encoding with CycPOEM (Cyclic Position Offset Encoding Matrix), a distance matrix that accounts for head-to-tail cyclization and disulfide bond shortcuts. The rest of the AlphaFold2/ColabFold pipeline — MSA generation, Evoformer attention, and structure module — remains unchanged. The result is substantially more accurate cyclic peptide structures without retraining any neural network weights.
The method was developed at Zhejiang University of Technology and Shanghai Highslab Therapeutics, published in Briefings in Bioinformatics (2024).
AlphaFold encodes the relative position between two residues as a simple linear offset: residue and residue are apart. For a cyclic peptide of length , this ignores that residues 1 and are covalently bonded.
CycPOEM constructs a more accurate distance matrix using a modified Floyd-Warshall shortest-path algorithm:
This means two residues on opposite sides of a 20-residue cyclic peptide are encoded as being 10 apart rather than 19 — reflecting the actual molecular topology.
CycPOEM also encodes directionality. The Upper Negative (UN) strategy, which assigns negative values to the upper triangle of the matrix, produced the best results in benchmarks. This captures the asymmetric nature of peptide bonds (N→C directionality).
On a test set of 63 NMR-resolved cyclic peptide structures, HighFold achieved a median backbone RMSD of 1.058 Å, compared to 1.737 Å for AfCycDesign and 1.956 Å for standard AlphaFold. For cyclic peptides with disulfide bridges, the improvement was even more pronounced: 1.720 Å average RMSD versus 3.256 Å for AfCycDesign.
ProteinIQ runs HighFold on A100 GPU infrastructure with pre-loaded AlphaFold2 model weights, eliminating the need to configure ColabFold, install HighFold's overlay, or manage GPU environments locally.
| Input | Description |
|---|---|
Cyclic peptide | Amino acid sequence in FASTA or raw format. Single chain only. Standard amino acids (20 canonical residues). |
| Setting | Description |
|---|---|
Disulfide bond pairs | Optional, explicitly known cysteine pairs, e.g. 1 5, 3 8. Residue numbers are 1-based, every referenced residue must be cysteine, and a cysteine can appear in only one pair. Leave empty for head-to-tail cyclization only. ProteinIQ does not infer unknown disulfide connectivity. |
| Setting | Default | Description |
|---|---|---|
Number of models | 5 | AlphaFold2 models to run (1–5). All five gives the most reliable ranking but increases runtime proportionally. |
Random seeds | 1 | Number of random seeds per model (1–5). More seeds produce greater diversity in predicted conformations. |
Recycles | 3 | Number of recycling iterations through the model (1–20). More recycles can improve accuracy for difficult targets but increases runtime. |
Model type | AlphaFold2-pTM | Model variant. AlphaFold2-pTM adds pTM confidence output. HighFold preserves the source model ranking by pLDDT for monomeric cyclic peptides. |
| Setting | Default | Description |
|---|---|---|
MSA mode | UniRef+Environmental | Database for multiple sequence alignment generation via MMseqs2. UniRef+Environmental searches UniRef and environmental sequence databases. UniRef only is faster. No MSA runs single-sequence mode (no evolutionary information). |
Use templates | On | Query the PDB for structural templates. Useful when homologous structures exist. |
Max MSA sequences | Auto | Maximum number of MSA sequences to use. Leave empty for automatic selection. Lower values speed up prediction at the cost of accuracy. |
| Setting | Default | Description |
|---|---|---|
AMBER relaxation | On | Run OpenMM/AMBER energy minimization on predicted structures. Recommended for cyclic peptides, where strained geometry is common. |
Structures to relax | 1 | Number of top-ranked predictions to relax (0–5). Only applies when AMBER relaxation is enabled. |
Early stop tolerance | Disabled | Stop recycling early if pLDDT improvement is below this threshold (e.g. 0.5). Speeds up prediction for easy targets. |
Random seed | 0 | Random seed for reproducibility. Use the same seed and settings to reproduce identical results. |
Each prediction run produces up to the selected model count multiplied by the selected seed count. Results preserve HighFold's source ranking and include:
.a3m format).png)| pLDDT | Interpretation |
|---|---|
| > 90 | High confidence — backbone and sidechain positions likely accurate |
| 70–90 | Good confidence — backbone probably correct, sidechains less certain |
| 50–70 | Low confidence — treat with caution, may need experimental validation |
| < 50 | Very low confidence — structure unreliable in this region |
For cyclic peptides, pLDDT values tend to be lower than for globular proteins of similar size, particularly in loop regions. A pLDDT of 70+ for a short cyclic peptide generally indicates a successful prediction.
No MSA mode for very short sequences.