ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
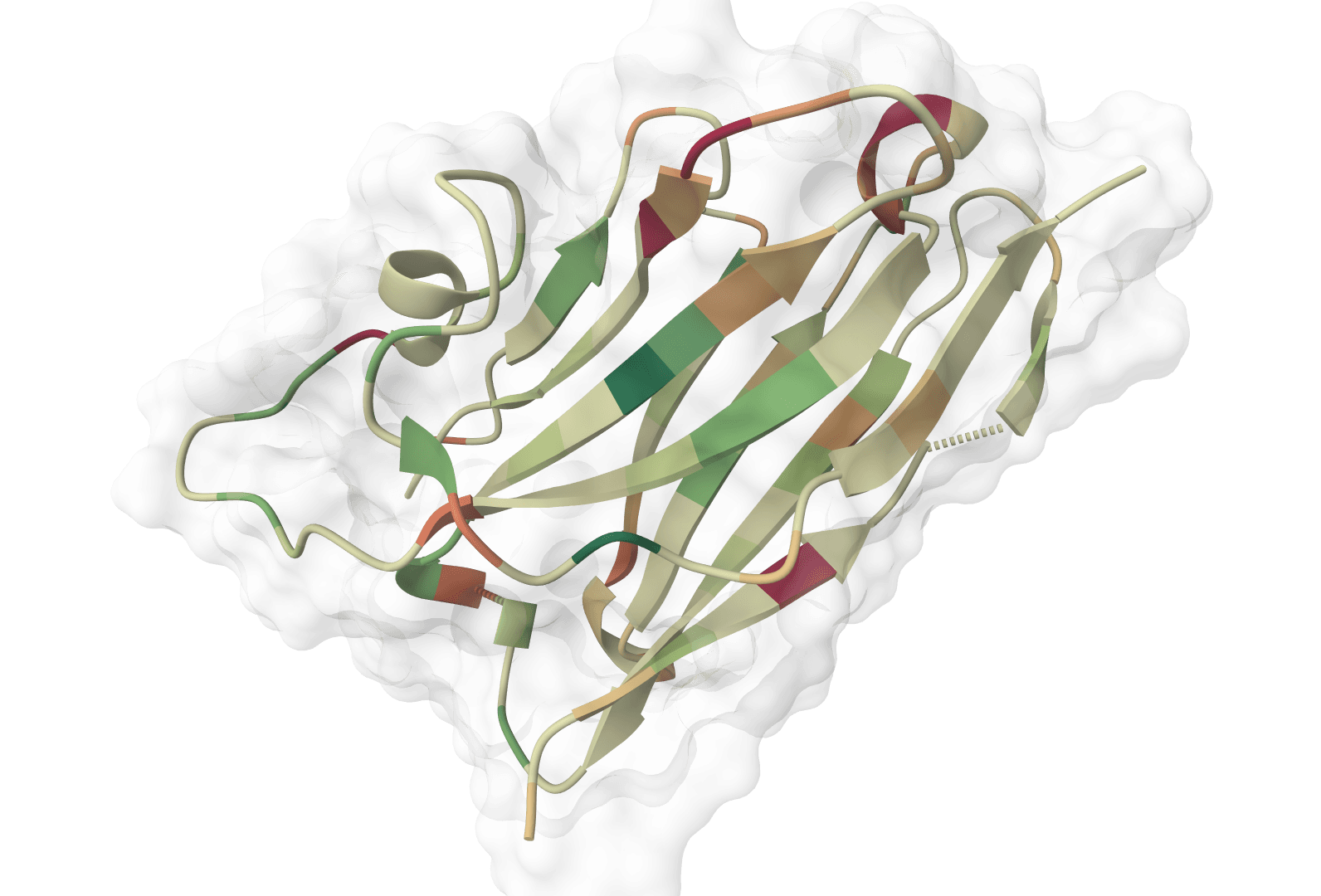
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
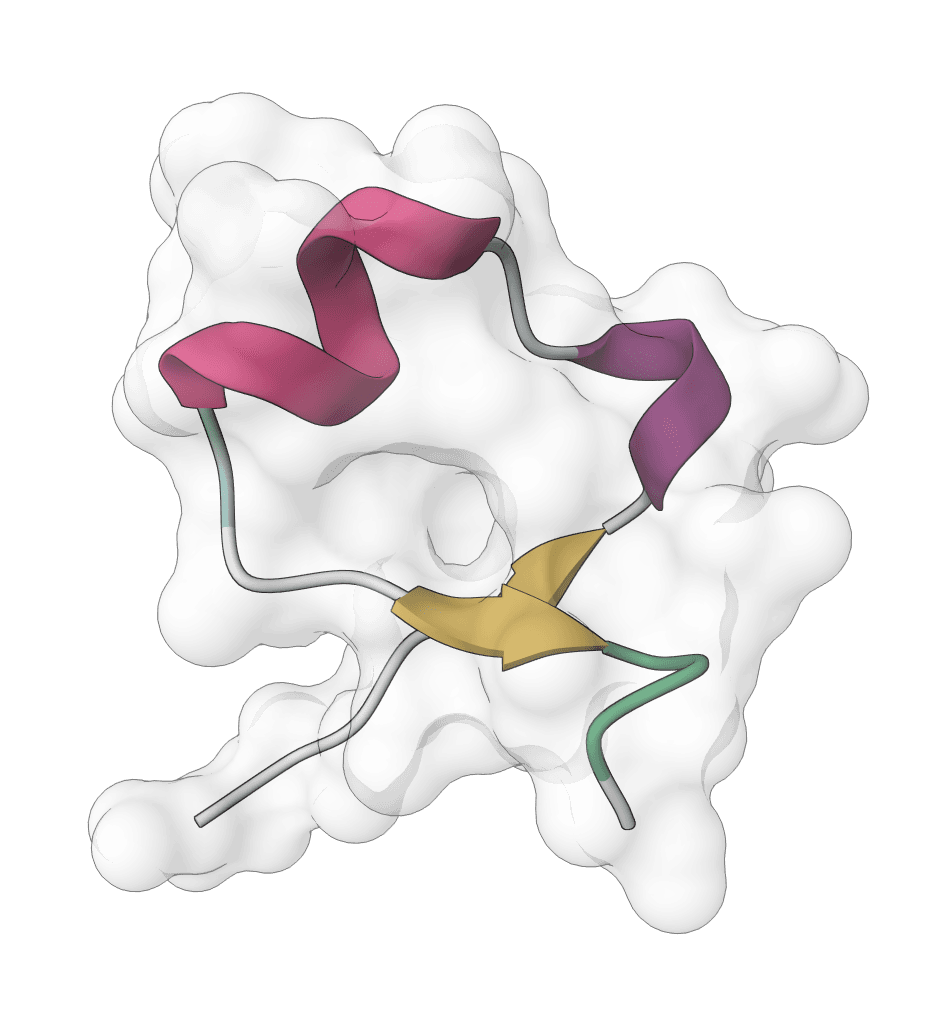
Controllable all-atom structure prediction for proteins, ligands, DNA, RNA, and multi-component complexes using IntelliFold 2.0.4 on its AlphaFold 3 JAX engine.
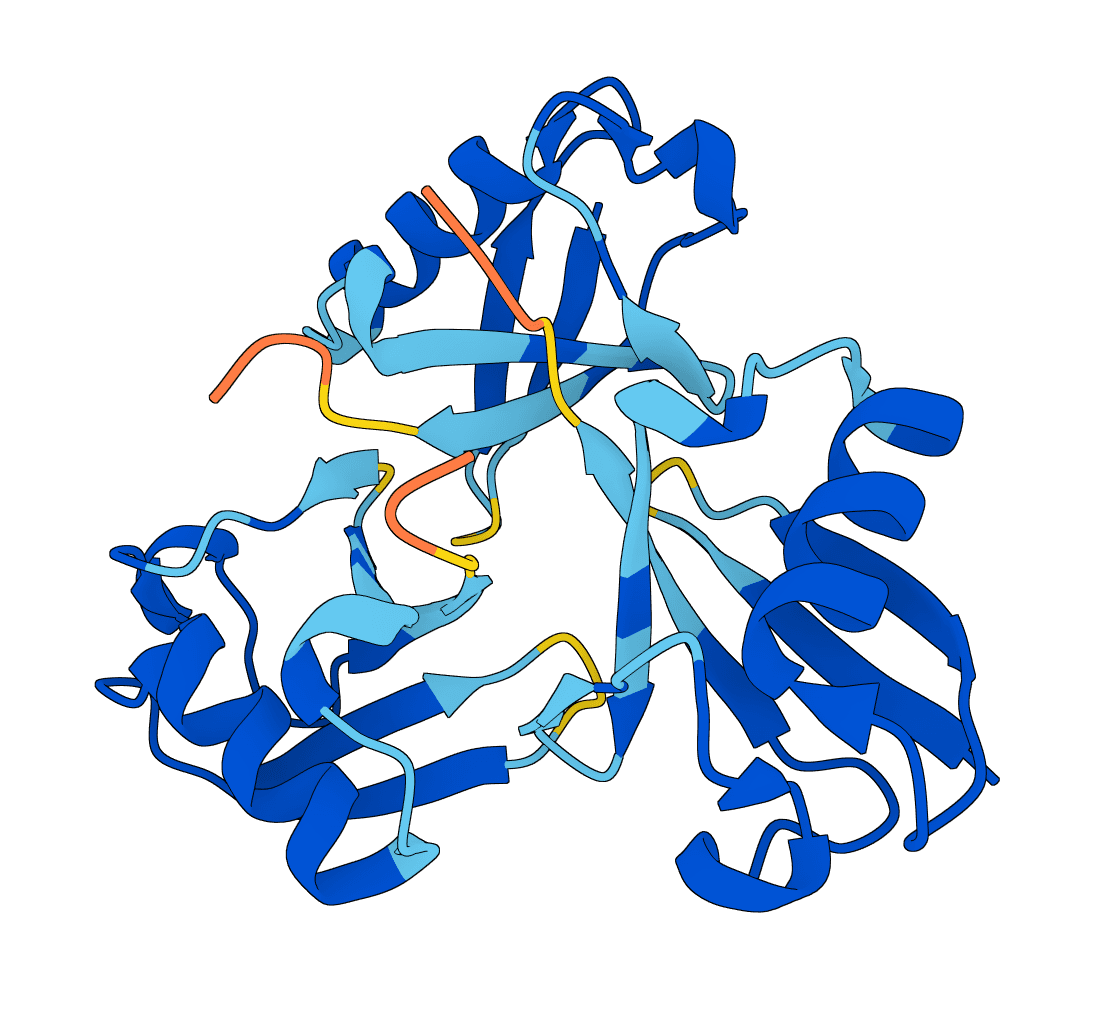
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Controllable all-atom structure prediction for proteins, ligands, DNA, RNA, and multi-component complexes using IntelliFold 2.0.4 on its AlphaFold 3 JAX engine.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
Configure inputs to begin
Set options on the left, then click “Submit job”.
MiniFold is a streamlined protein structure prediction model that delivers ESMFold-level accuracy at 10–20 times the speed. Developed for large-scale applications, MiniFold predicts three-dimensional protein structures from single amino acid sequences without requiring multiple sequence alignments (MSAs).
The model achieves its exceptional efficiency through a redesigned Evoformer architecture—a simplified version of the component introduced in AlphaFold2—paired with a lightweight structure module and custom GPU kernels optimized for protein folding tasks.
Traditional folding models like AlphaFold2 achieve high accuracy through computationally expensive operations on large MSAs and complex structural refinement modules. ESMFold eliminated the MSA requirement by learning from a protein language model (ESM-2), but retained a relatively heavy Evoformer stack for processing evolutionary information.
MiniFold builds on ESM-2 like ESMFold does, but streamlines both the Evoformer and structure module. The redesigned Evoformer reduces computational overhead while preserving the model's ability to capture long-range dependencies in protein sequences. Custom GPU kernels exploit parallelism at the hardware level, cutting both inference time and peak memory consumption by substantial margins.
The result: predictions that complete in seconds rather than minutes, with benchmark accuracy matching ESMFold on CAMEO and CASP datasets.
ProteinIQ runs MiniFold on GPU infrastructure, delivering structure predictions directly in your browser with no installation required.
| Input | Description |
|---|---|
Protein sequence | Exactly one FASTA record or raw amino acid sequence. Accepts the 20 standard amino-acid codes plus X; RCSB selections are resolved to sequence content before submission. |
MiniFold offers two model variants and configurable recycling to balance speed and accuracy:
| Setting | Options | Description |
|---|---|---|
Model size | 48L (default), 12L | Number of layers in the Evoformer. 48L provides better accuracy but takes longer. 12L is faster with minimal accuracy loss for well-characterized protein families. |
Recycling iterations | 1–6 (default: 3) | Number of MiniFold recycling iterations. Higher values may improve quality for difficult sequences but increase computation time. |
MiniFold returns its generated structure in PDB format:
MiniFold inherits pLDDT (predicted local distance difference test) scoring from AlphaFold:
| pLDDT range | Interpretation |
|---|---|
| > 90 | Very high confidence; backbone geometry and side-chain positions likely accurate |
| 70–90 | Generally reliable; backbone probably correct, some side-chain uncertainty |
| 50–70 | Low confidence; treat with caution, especially for functional site annotation |
| < 50 | Very low confidence; region may be intrinsically disordered or beyond model capability |
Regions with low pLDDT often correspond to flexible loops, disordered tails, or structural motifs lacking representation in the training data.
Use MiniFold when:
Use ESMFold when:
Use AlphaFold 2 when:
Use Chai-1 or Boltz-2 when:
MiniFold inherits the single-sequence limitation of ESMFold: without MSA information, it relies entirely on patterns learned during training. This works well for proteins with homologs in the training set, but struggles with:
For sequences outside these constraints, MSA-based predictors like AlphaFold 2 generally outperform single-sequence models. For a related single-sequence folding option, compare with ESMFold2.