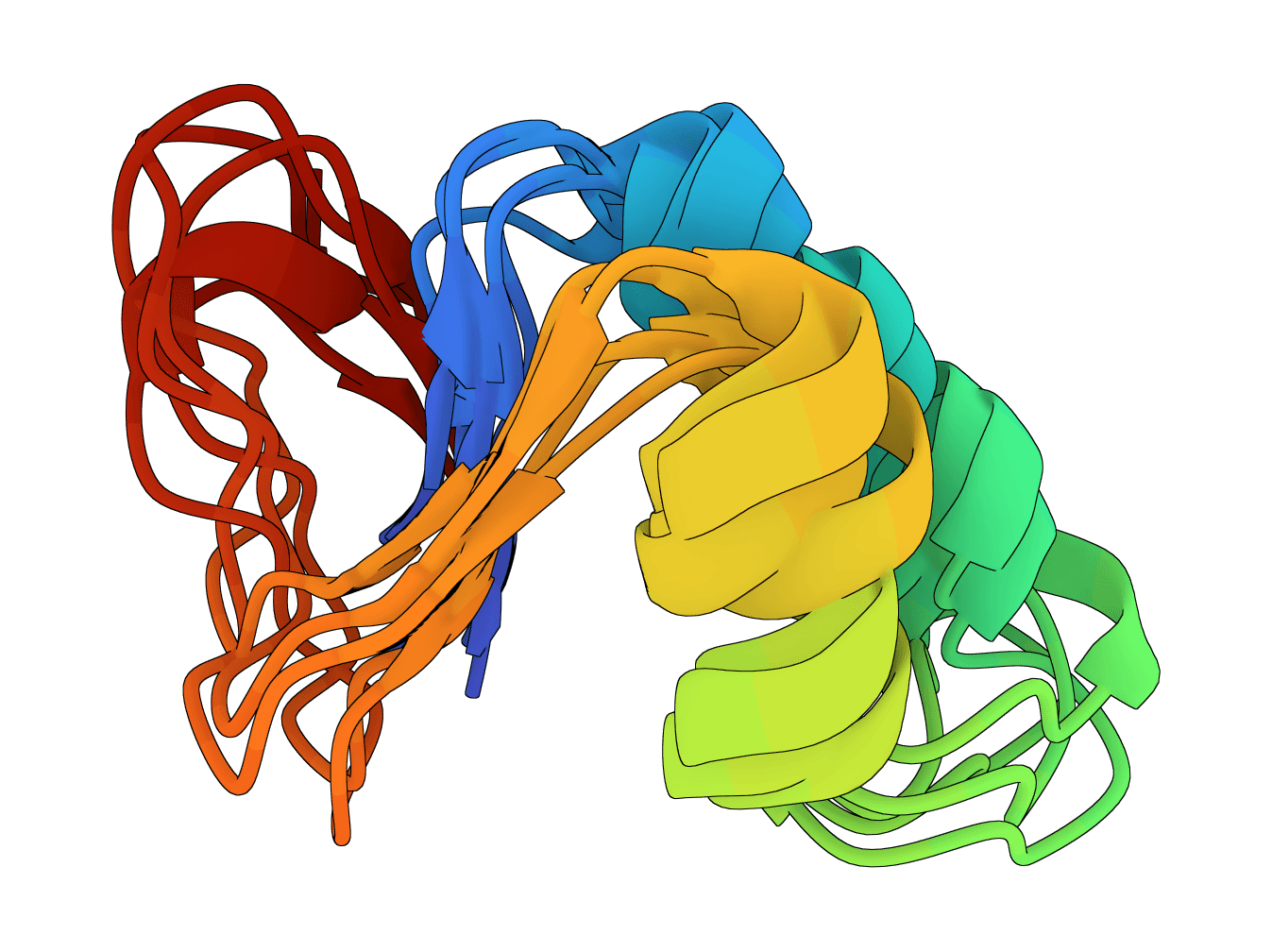
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Controllable all-atom structure prediction for proteins, ligands, DNA, RNA, and multi-component complexes using IntelliFold 2.0.4 on its AlphaFold 3 JAX engine.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
MiniFold is a fast single-sequence protein structure predictor that is 10-20x faster than ESMFold. It predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it ideal for rapid structure prediction.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Controllable all-atom structure prediction for proteins, ligands, DNA, RNA, and multi-component complexes using IntelliFold 2.0.4 on its AlphaFold 3 JAX engine.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
MiniFold is a fast single-sequence protein structure predictor that is 10-20x faster than ESMFold. It predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it ideal for rapid structure prediction.
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Human Ubiquitin (76 aa) - Flexible regions
Protein structures in the PDB and in molecular dynamics simulations rarely describe a single rigid shape. Loops open and close, termini wander, domains breathe, and binding sites can appear only in a subset of conformations. AlphaFlow models that variability by turning AlphaFold-style structure prediction into a generative ensemble method.
AlphaFlow, developed by Jing, Berger, and Jaakkola, includes two model families. AlphaFlow uses AlphaFold with multiple sequence alignments. ESMFlow uses ESMFold and runs from sequence alone. ProteinIQ supports both model families: ESMFlow for single-sequence ensemble generation, and AlphaFlow for MSA-backed AlphaFold-style ensemble generation.
For a single best-guess structure, ESMFold or AlphaFold 2 is usually the clearer choice. AlphaFlow is useful when the question is about plausible conformational spread rather than one static model.
AlphaFlow runs online in ProteinIQ by selecting an input mode, providing one protein sequence, choosing a matching model variant, and generating a conformational ensemble as downloadable PDB files. ESMFlow mode needs only a sequence. AlphaFlow mode accepts the same sequence plus an optional .a3m multiple sequence alignment, and generates the alignment automatically when none is uploaded.
| Input | Accepted format | Notes |
|---|---|---|
Protein sequence | FASTA, raw amino acid sequence, .fasta, .fa, or .txt | Exactly one protein sequence between 10 and 1600 residues. Ambiguous alphabetic residue codes are preserved as X; unsupported non-letter characters are rejected. |
Pre-computed MSA | Optional .a3m | Appears only after AlphaFlow input mode is set to AlphaFlow. The uploaded alignment must be valid A3M with the query sequence first. Invalid .a3m files fail clearly rather than falling back to generated MSA. |
RCSB fetch | PDB ID, for example 1UBQ | ProteinIQ fetches FASTA from RCSB when a PDB ID is used. The entry must resolve to exactly one FASTA record; the resulting sequence, not the PDB coordinates, is used for inference. |
Job name | Text | Optional label for finding the run later. |
AlphaFlow on ProteinIQ is designed for single-chain protein sequences. Multimer interfaces, ligand-bound states, membranes, cofactors, and chain-specific contacts are not modeled directly from the input. When a PDB ID is fetched from RCSB, the sequence is used as the input and the deposited coordinates are not used as a structural template.
Uploaded .a3m alignments are checked before the job starts. ProteinIQ accepts permissive A3M content, but rejects files that would make the AlphaFold-style feature pipeline ambiguous.
| Check | Requirement |
|---|---|
| FASTA headers | Each record starts with a non-empty header line beginning with >. |
| Sequence records | Every header has at least one sequence line. |
| Characters | Sequences use source-compatible A3M characters: amino acid letters, lowercase insertion letters, or -. |
| Aligned length | After stripping lowercase insertion characters, every record has the same aligned column length. |
| Query sequence | The first record has a non-empty ungapped query sequence and exactly matches the normalized submitted protein after lowercase insertions and gaps are removed. Use X at ambiguous residue positions. |
If no .a3m is uploaded in AlphaFlow mode, ProteinIQ generates an MSA server-side with an MMseqs2 workflow compatible with ColabFold-style A3M output. The generated alignment is returned with the results so the run can be inspected or repeated.
X, B, Z, or U are mapped to X without changing sequence positions. Resolve them before inference when possible.Human ubiquitin (1UBQ, 76 residues) is a useful sanity-check target because the beta-grasp fold should remain compact while termini and surface loops vary modestly across samples. A run where the core unfolds or most samples look unrelated is a warning sign to inspect the input sequence and settings.
| Setting | Default | Description |
|---|---|---|
AlphaFlow input mode | ESMFlow | Controls both the input form and the available model variants. ESMFlow uses sequence only. AlphaFlow shows the optional .a3m upload and otherwise generates an MSA server-side. |
Model variant | ESMFlow MD Base in ESMFlow mode, AlphaFlow MD Base in AlphaFlow mode | Selects the checkpoint. The menu is scoped to the selected input mode, so ESMFlow variants appear only in ESMFlow mode and AlphaFlow variants appear only in AlphaFlow mode. |
Number of samples | 10 | Number of PDB conformations to generate, from 1 to 50. More samples give a better view of ensemble spread, but runtime scales roughly linearly with sample count. |
MD models learn conformational variation from molecular dynamics trajectories. PDB models learn diversity across experimental structures. Base models are the full checkpoints. Distilled models are faster and automatically use the distilled inference settings.
| Variant | Best fit | Practical tradeoff |
|---|---|---|
ESMFlow MD Base | Studying thermal flexibility, mobile loops, termini, and domain breathing around physiological conditions | Recommended default. Slower than distilled models, but closest to the full MD-trained ESMFlow behavior. |
ESMFlow PDB Base | Looking for experimental-state diversity, such as alternative crystal forms or condition-dependent conformations | Can reflect broader structural heterogeneity than MD-like local motion. |
ESMFlow MD Distilled | Fast exploratory ensemble generation when many sequences need screening | Faster, with some accuracy loss relative to the base model. Uses No diffusion and Noisy first automatically. |
ESMFlow PDB Distilled | Fast comparison of experimental-ensemble-like conformations | Useful for triage, not the best choice for final structural interpretation. |
AlphaFlow MD Base | MSA-backed molecular-dynamics-like ensembles | Uses AlphaFold-style MSA features. Slower and more expensive than ESMFlow, but closer to the original AlphaFlow method. |
AlphaFlow MD Distilled | Faster MSA-backed MD-like exploratory ensembles | Uses the distilled inference mode. |
AlphaFlow PDB Base | MSA-backed experimental-state ensemble diversity | Enables Self conditioning and MSA resampling by default for PDB-style sampling. |
AlphaFlow PDB Distilled | Faster MSA-backed PDB-like ensembles | Uses PDB-style sampling plus the distilled inference mode. |
| Setting | Default | Description |
|---|---|---|
Inference steps | 10 | Number of denoising steps, from 2 to 50. The standard setting is 10. Lower values truncate generation and can reduce diversity. Higher values cost more runtime. |
Diversity (tmax) | 1.0 | Starting point in the generation schedule, from 0.2 to 1.0. 1.0 samples from the full schedule. Lower values keep samples closer together. |
Self conditioning | Off | Enables --self_cond. AlphaFlow PDB variants enable it automatically for PDB-style sampling. |
No diffusion | Off | Enables --no_diffusion. Distilled variants enable this automatically. |
Noisy first | Off | Enables --noisy_first. Distilled variants enable this automatically. |
The two controls that most users should change first are Number of samples and Model variant. Inference steps, Diversity (tmax), and the boolean inference flags are better treated as reproducibility controls. Changing them changes the sampling procedure, not just the display.
Both ESMFlow and AlphaFlow return the native AlphaFlow multi-model PDB as the primary result. ProteinIQ also splits ensembles within the derived-output size budget into secondary per-sample PDB files for convenient viewing; very large ensembles remain available in their complete native multi-model file without duplicating the payload. AlphaFlow mode returns the uploaded or generated .a3m, and both modes return source runtime data and the source-entrypoint log when available.
| Output | Meaning |
|---|---|
{name}.pdb | Primary native multi-model PDB containing the complete sampled ensemble. |
sample_01.pdb, sample_02.pdb, ... | Secondary individual conformations split from the native ensemble for convenient viewing and download. |
{name}.a3m | AlphaFlow modes only. Uploaded or generated MSA used for inference. |
{name}_runtime.json | Per-sample inference runtimes reported by the pinned AlphaFlow entrypoint. |
{name}_alphaflow.log | Standard output and error text from the AlphaFlow source entrypoint. |
Model variant | Checkpoint used for the run, such as esmflow_md_base or esmflow_pdb_distilled. |
Sample index | Position of the sampled conformation in the generated ensemble. It is not a confidence rank. |
Inference settings | steps, tmax, self_cond, no_diffusion, and noisy_first are recorded so runs can be compared later. |
AlphaFlow does not return a pLDDT confidence table or a binding score. The value of the run is in the ensemble itself: how similar the structures are, which regions move, and whether the sampled conformations suggest more than one plausible state.
Results record the reviewed AlphaFlow source commit 796971961368c9bd1504b3d57c5348a3d1d4ecaa, the predict.py entrypoint, the immutable model-repository revision, and the selected checkpoint checksum.
| Pattern in the ensemble | Likely interpretation | Follow-up check |
|---|---|---|
| Core secondary structure overlaps across most samples | The model has a consistent fold hypothesis | Inspect loops, termini, and domain interfaces rather than whole-protein RMSD alone. |
| One loop repeatedly opens or closes | The loop may be mobile or weakly constrained by sequence context | Compare with known active-site loops, disorder predictions, or mutagenesis data. |
| Two domains stay folded but change relative orientation | Hinge-like motion may be plausible | Align one domain first, then measure displacement of the other domain. |
| Only one sample shows an extreme rearrangement | The structure is more likely an outlier than a stable alternate state | Look for the same movement in additional samples before using it for docking or design. |
| Most samples are globally inconsistent | The input may be too long, disordered, or outside the model's reliable regime | Try domain-level constructs or compare a single-structure prediction with ESMFold. |
An AlphaFlow output should be read as a set of plausible conformations, not as a ranked list where sample 1 is best. The sample index only records generation order.
Large differences across samples usually point to regions where the model sees conformational uncertainty or mobility. In practice, the most useful checks are visual:
Quantitative RMSD analysis is useful after the job, but RMSD needs a reference choice. Whole-protein RMSD can be dominated by flexible tails. For many proteins, aligning the stable core first and then measuring loop or domain movement gives a clearer interpretation.
AlphaFlow trains structure predictors as flow-matching models. During training, the model sees an interpolation between a noisy protein backbone representation and a target structure:
Here, is sampled from a harmonic prior, is the target structure, and marks progress along the denoising path. At inference time, the model starts from a noisy configuration and repeatedly predicts a cleaner structure along the schedule.
ESMFlow applies the same idea to ESMFold. Because ESMFold is a single-sequence model, ESMFlow can run without MSA generation. That makes it practical for fast web execution and for proteins where homologous sequence coverage is weak.
AlphaFlow mode uses AlphaFold-style MSA features. A supplied .a3m file is used directly after validation. Without an upload, ProteinIQ generates the alignment first, then writes it into the directory layout expected by the AlphaFlow inference code before sampling structures. That extra MSA step is the main reason AlphaFlow mode is slower than ESMFlow mode.
| Training set | What it captures | Interpretation consequence |
|---|---|---|
MD | Molecular dynamics trajectories from ATLAS at 300K | Better for local thermal motion, loop mobility, and dynamic fluctuations around an existing fold. |
PDB | Experimental structures deposited under different conditions | Better for alternative crystallographic or cryo-EM states, ligand-bound differences, and broader experimental heterogeneity. |
The distinction matters. An MD-trained model may emphasize physically local fluctuations. A PDB-trained model may sample larger state changes if similar state diversity appears in experimental structures.
| Tool | Best use | Difference from AlphaFlow |
|---|---|---|
| AlphaFlow | Generating a conformational ensemble from one protein sequence | Produces multiple plausible structures rather than one best structure. |
| ESMFold | Fast single-structure prediction without MSA generation | Better when a single model and pLDDT-style confidence are needed. |
| AlphaFold 2 | High-accuracy structure prediction where MSA information is helpful | Usually stronger for a single final structure, but not designed to sample an ensemble. |
| AF-Cluster | Exploring alternate AlphaFold conformations by clustering subsampled MSA predictions | Useful when MSA diversity may reveal different states, but it still depends on AlphaFold-style prediction runs. |
| MDGen | Generating protein conformations with a learned molecular dynamics model | Better when the goal is learned conformational sampling rather than AlphaFold-style ensemble prediction. |
| Boltz-2 | Structure prediction for complexes involving proteins, ligands, DNA, or RNA | Better for multimolecular complexes and binding affinity workflows. |
| MD trajectory analysis | Measuring flexibility from an existing molecular dynamics trajectory | Works from simulated trajectory data instead of generating conformations from sequence. |
AlphaFlow fits best between structure prediction and simulation. It can suggest flexible regions worth checking before setting up molecular dynamics, docking, mutagenesis, or construct design. It should not replace experimental dynamics data when NMR, HDX-MS, cryo-EM heterogeneity, or high-quality MD trajectories are available.
.a3m files make runs more reproducible; automatic MMseqs2 generation depends on external search availability.