SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
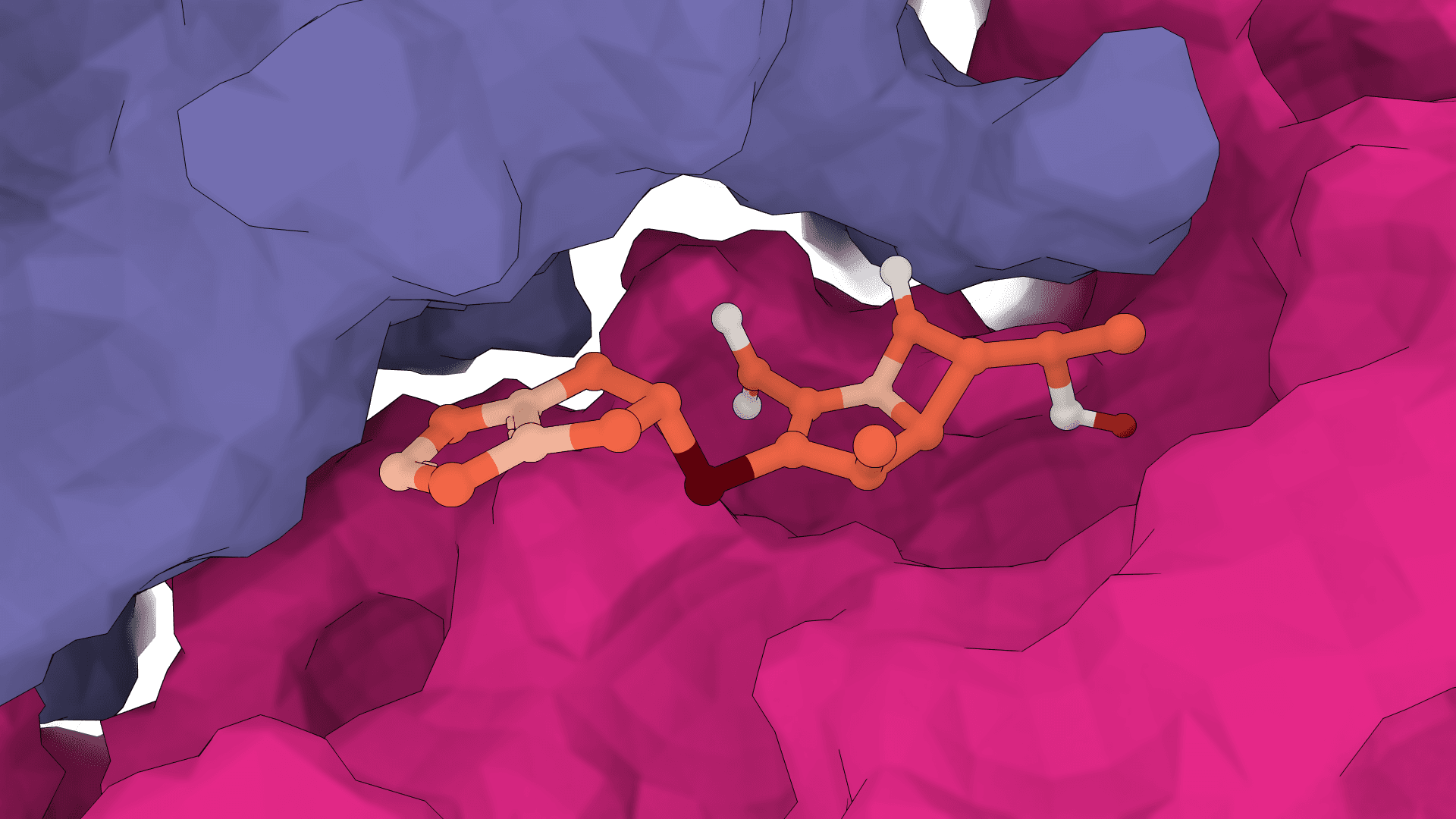
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
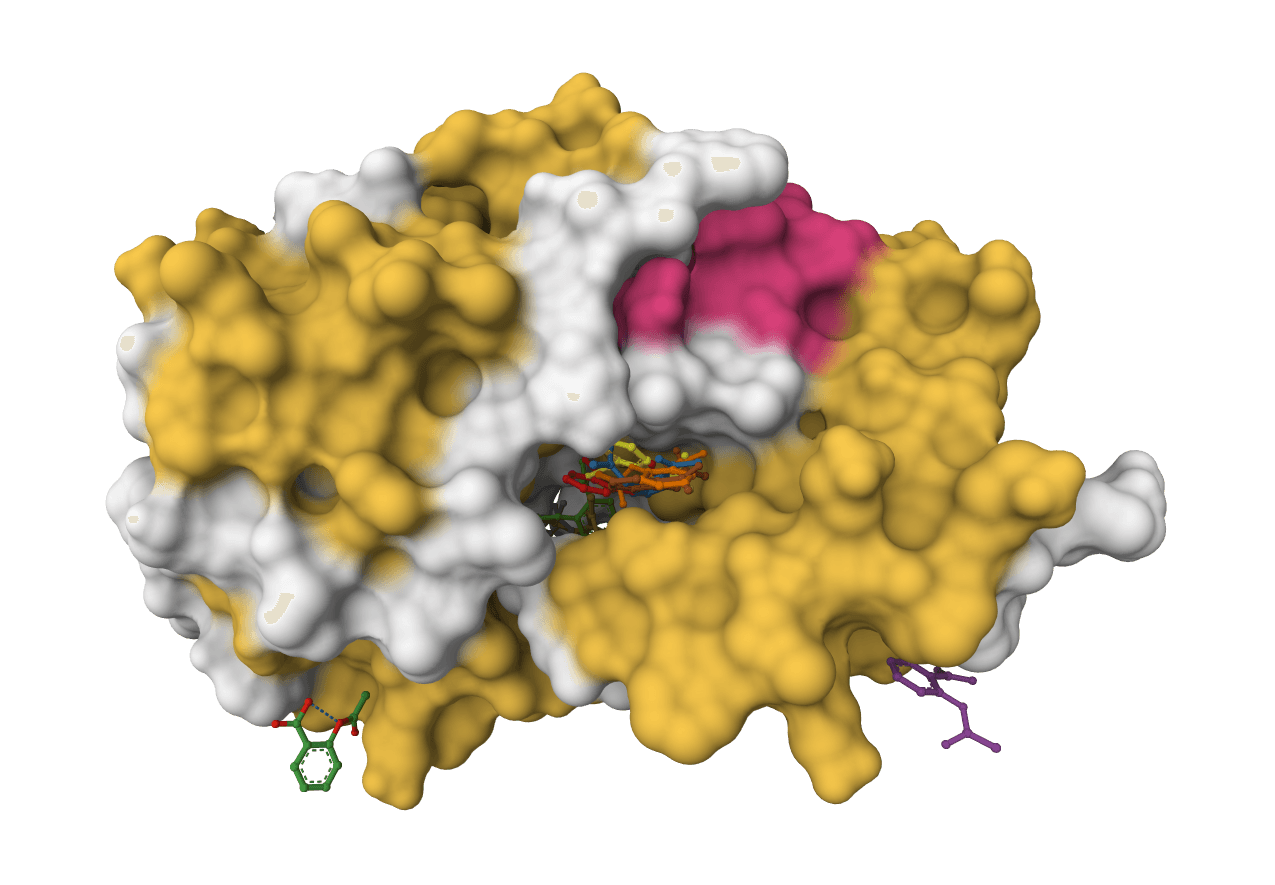
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
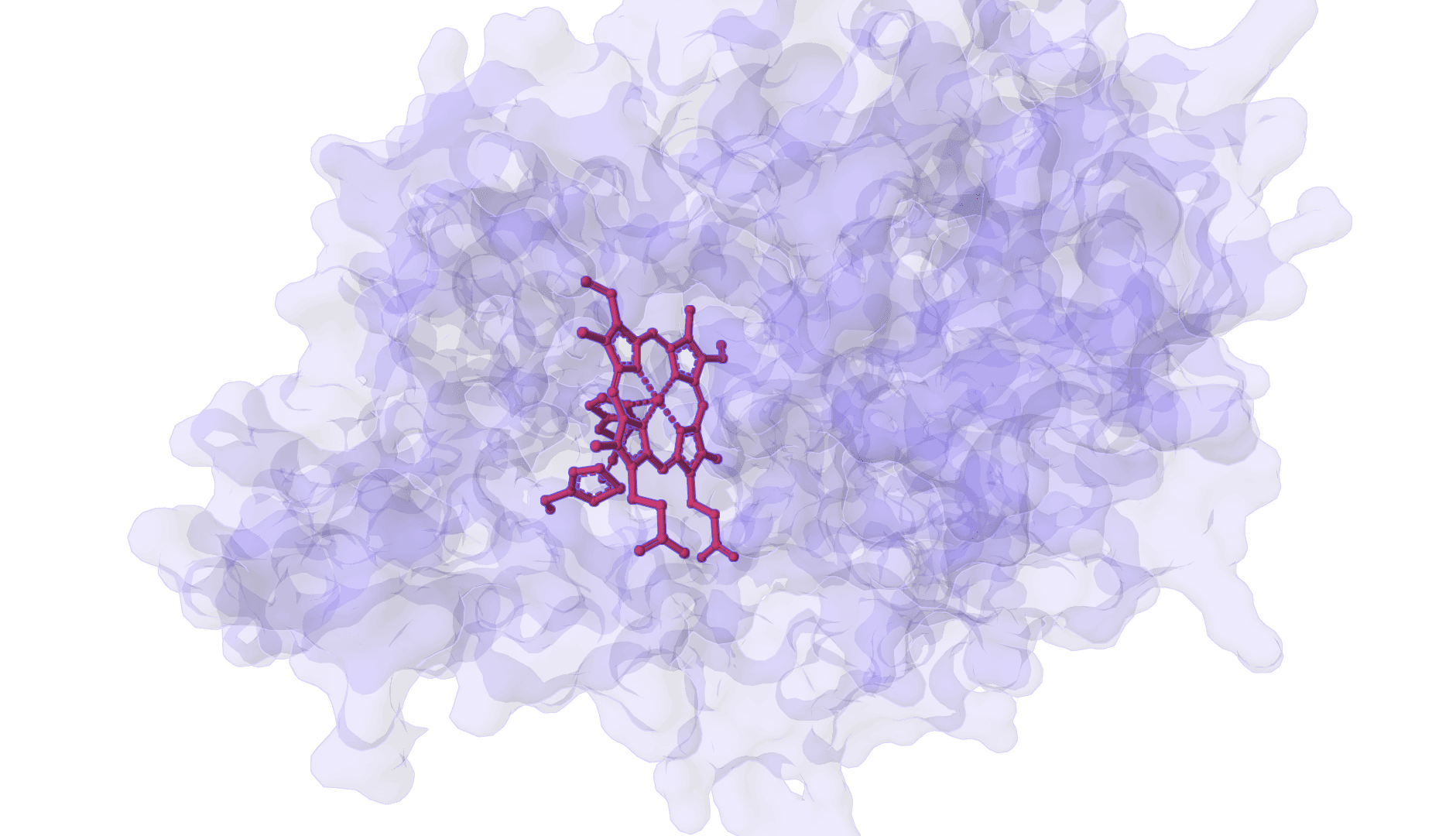
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
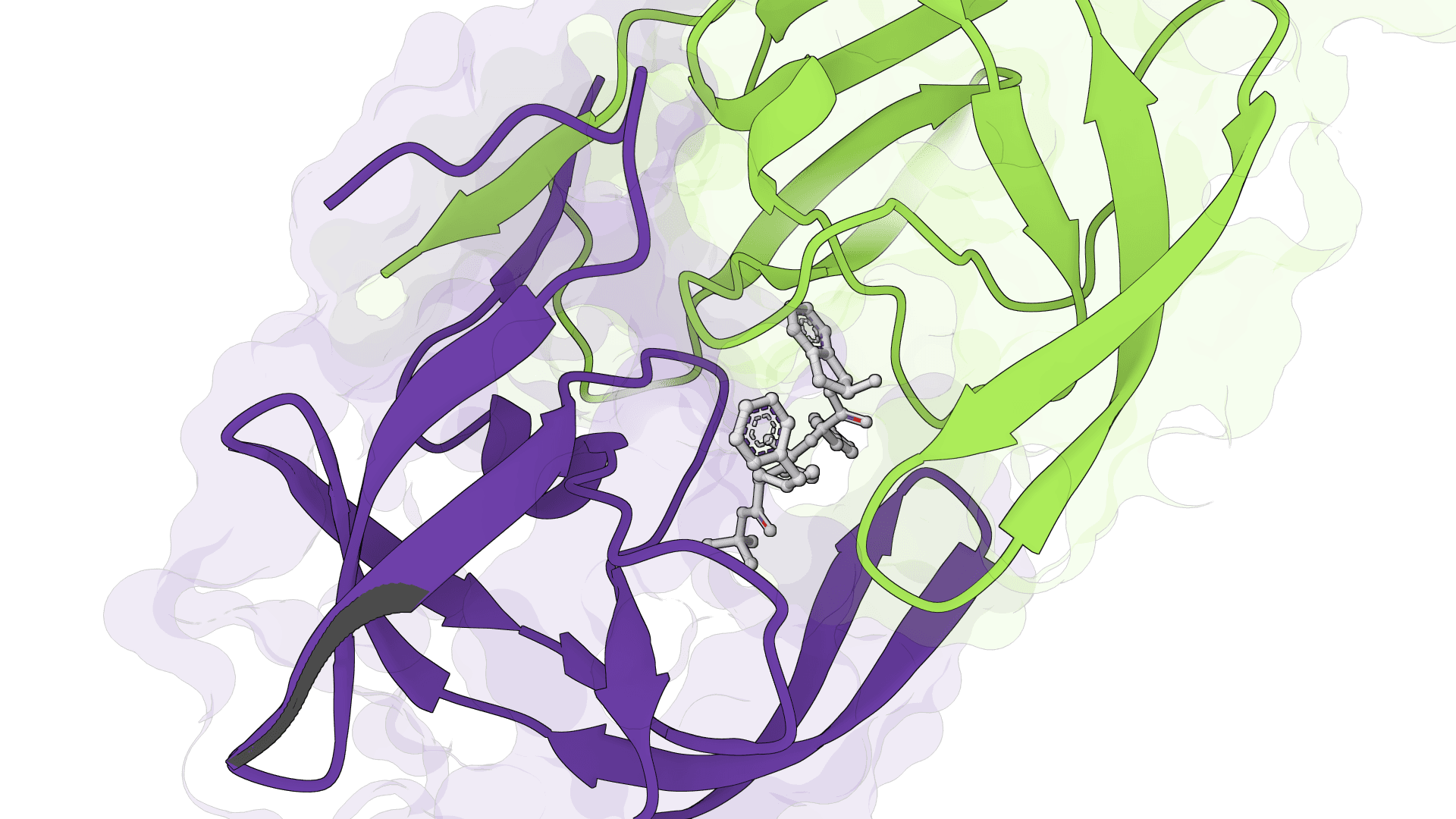
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
FlowDock predicts protein-ligand complex structures and binding-affinity scores using geometric flow matching.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
Configure inputs to begin
Set options on the left, then click “Submit job”.
PandaDock is an open-source molecular docking program written in Python. It predicts how small-molecule ligands bind to protein targets by searching for low-energy binding poses and scoring them with physics-based or hybrid energy functions. Developed by Pritam Kumar Panda, PandaDock is designed entirely around CPU computation — no GPU is required — and achieves sub-angstrom structural accuracy on standard benchmarks (0.014 Å mean RMSD on PDBbind v2020 complexes, with 99.3% of poses within 2 Å of the crystallographic reference).
Where most docking tools offer a single search strategy, PandaDock bundles several algorithms under one interface. An enhanced hierarchical search (the default) runs a coarse global scan, intermediate refinement, and fine local optimization in three stages. Monte Carlo sampling uses simulated annealing. A genetic algorithm applies crossover and mutation operators to evolve pose populations. A crystal-guided mode constrains the search near a known binding geometry for re-docking validation. Each algorithm pairs with any of the available scoring functions.
PandaDock's docking workflow follows three phases: search space definition, conformational sampling, and scoring/ranking.
If no binding site is specified, PandaDock computes a bounding box around all protein atoms with padding. When the binding pocket is known, coordinates and box dimensions can be set manually to focus the search and reduce computation.
The default Enhanced Hierarchical CPU algorithm operates in three stages:
Alternative algorithms trade off speed and thoroughness differently. Monte Carlo sampling explores conformational space through random perturbations accepted or rejected by a Boltzmann criterion, making it faster for screening. The genetic algorithm maintains a population of poses that evolve over generations — useful for complex binding sites with multiple local minima.
| Function | Description |
|---|---|
Physics-based | Full force-field evaluation including van der Waals, electrostatics, hydrogen bonding, and desolvation terms. Best general-purpose accuracy. |
Empirical | Statistical potential derived from known protein-ligand complexes. Faster, suited for rapid screening. |
Precision Score | Higher-resolution energy calculation with tighter convergence criteria. Slower but more discriminating for closely ranked poses. |
Hybrid | Combines physics-based energy terms with machine-learning rescoring for improved affinity ranking. |
ProteinIQ hosts PandaDock on cloud infrastructure so docking jobs can be submitted directly from the browser — no Python environment, dependencies, or command-line setup needed.
| Input | Description |
|---|---|
Protein (Receptor) | PDB file upload or 4-character PDB ID (e.g., 1HSG). The structure is fetched from RCSB if an ID is provided. |
Ligand | SMILES string, SDF/MOL file, or PubChem compound name. SMILES are converted to 3D coordinates automatically using RDKit. |
| Setting | Description |
|---|---|
Docking algorithm | Search strategy. Enhanced Hierarchical CPU (default, recommended) runs a three-stage search for maximum accuracy. Monte Carlo CPU is faster for screening. Genetic Algorithm CPU handles complex binding sites. Crystal-Guided CPU is for re-docking validation. Auto selects based on input characteristics. |
Scoring function | Energy evaluation method. Physics-based (default) for general use, Empirical for speed, Precision Score for fine discrimination, Hybrid for ML-enhanced ranking. |
Number of poses | How many binding poses to generate (1–20, default 5). More poses sample more of the energy landscape at the cost of longer runtime. |
Manual search space configuration is available for users who know the binding pocket location. When disabled (default), PandaDock automatically computes a box enclosing the entire protein.
| Setting | Description |
|---|---|
Configure search space | Toggle manual binding site specification. |
Center X/Y/Z | Coordinates of the search box center in Angstroms. |
Box size X/Y/Z | Dimensions of the search box (5–100 Å, default 22.5 Å per axis). |
PandaDock returns ranked binding poses viewable in an interactive 3D viewer alongside downloadable files.
| Output | Description |
|---|---|
| Ligand poses | Individual ligand conformations (PDB format), ranked by score. Displayed in the 3D viewer superimposed on the receptor. |
| Complex structures | Full protein-ligand complexes for each pose, available for download. |
| Visualization plots | Binding affinity and interaction energy charts (PNG). |
| Interaction analysis | Hydrogen bonds, hydrophobic contacts, and energy decomposition (JSON). |
PandaDock scores are unitless energy-like values where lower is better — a more negative score indicates a more favorable predicted binding interaction. Scores are most meaningful when comparing poses within the same docking run rather than across different protein-ligand pairs.
| Score range | Interpretation |
|---|---|
| Most negative (rank 1) | Predicted best binding pose |
| Near zero | Weak or unfavorable interaction |
Because PandaDock uses its own scoring function rather than calibrated free-energy estimates, scores should not be interpreted as binding affinities in kcal/mol. For absolute affinity prediction, consider re-scoring top poses with dedicated tools or running experimental validation.