Calculate RMSD, RMSF, and Rg from MD trajectories Learn more
Run
1 credit
Output
Configure inputs to begin
Set options on the left, then click “Submit job”.
Calculate RMSD, RMSF, and Rg from MD trajectories Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
Calculate RMSD, RMSF, and Rg from MD trajectories Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
MD trajectory analysis turns a simulation trajectory into measurements that can actually be interpreted: structural drift, residue flexibility, compactness, contact persistence, secondary-structure content, collective motions, and solvent-facing behavior. The raw frames coming out of GROMACS, AMBER, CHARMM, NAMD, or OpenMM do not answer those questions on their own.
This ProteinIQ tool runs a workflow pinned to MDAnalysis 2.9.0. It is not a single command-line program. It is a curated analysis surface built from MDAnalysis modules and a small amount of integration logic that standardizes inputs, runs multiple observables in one job, and returns structured outputs for charting and download.
The practical value is in combining observables that answer different failure modes:
Upload a topology file and a matching trajectory file, choose one or more analyses, and run the job to receive tabular and chart-ready outputs for each selected observable. The result page returns structural, interaction, conformational, environmental, and secondary-structure summaries with downloadable data for follow-up analysis.
| Input | Description | Accepted formats | Max size |
|---|---|---|---|
Topology/Structure file | Defines atom identities, residue assignments, connectivity, and metadata needed to interpret trajectory coordinates. | .pdb, .ent, .gro, .crd, .psf, .prmtop, .top, .tpr | 50 MB |
Trajectory file | Stores coordinates for each sampled frame. The topology and trajectory must describe the same system and atom ordering. | .xtc, .trr, .dcd, .nc, .ncdf, .tng, .crd, .xyz, .mdcrd | 500 MB |
Common pairings include:
.gro + .xtc for GROMACS.tpr + .xtc when topology metadata from GROMACS should be preserved.prmtop + .nc, .ncdf, or .mdcrd for AMBER.psf + .dcd for CHARMM or NAMD.pdb, .crd, or .gro with coordinate trajectories such as .xyz when atom ordering matchesThe tool loads the uploaded files into an MDAnalysis Universe, applies the selected atom selection, resolves the reference mode, optionally aligns sampled frames to that reference, and then dispatches the requested observables.
Three details matter because they change interpretation:
first, last, and average are not cosmetic options. average computes an average reference over the sampled frames instead of aliasing to the first frame.step subsamples the trajectory globally for the audited analysis paths, which reduces runtime and directly changes temporal resolution.Some analyses rely on additional assumptions from the underlying MDAnalysis modules or the tool:
3.0 Å and angle cutoff of 150°, matching the audited MDAnalysis default. Explicit hydrogens are still required for meaningful hydrogen-bond counts.protein and name CA selection is useful for fold tracking but does not contain hydrogens, so lifetime analysis will usually return a warning unless a chemically complete selection is used.MDAnalysis.analysis.contacts.Contacts and reports the source native-contact fraction Q. The displayed contact count is a compatibility summary derived from that fraction and the first-frame native contact set.MDAnalysis.analysis.msd.EinsteinMSD with msd_type="xyz". Physical diffusion estimates require trajectories in an unwrapped coordinate convention.MDAnalysis.analysis.dssp when available. If that path fails, the tool falls back to a simpler internal estimate rather than aborting the whole job.χ1/χ2 summaries use MDAnalysis Janin analysis with circular angle statistics. Helix geometry uses MDAnalysis HELANAL on alpha-carbon selections and requires consecutive helix segments of at least 9 residues.| Setting | Description | Default |
|---|---|---|
Analyses to run | Selects which observables are calculated. Multiple analyses can run in one job. | RMSD, RMSF, Radius of gyration |
Atom selection | MDAnalysis selection string applied to the primary analysis group. Examples include protein, protein and name CA, backbone, and resid 1-100. | protein and name CA |
Reference frame | Reference for RMSD-aligned analyses. Average structure computes the mean coordinate set over the sampled frames. | first |
| Setting | Description | Default |
|---|---|---|
Frame step | Samples every nth frame. A value of 10 processes frames 0, 10, 20, ... instead of every saved frame. | 1 |
Align trajectory | Aligns sampled frames to the chosen reference before RMSD, RMSF, PCA, clustering, and cross-correlation analysis. | true |
Second atom selection | Secondary MDAnalysis selection used for distance tracking and RDF. | resname SOL and name OW |
PCA components | Number of principal components returned in PCA projections. | 3 |
Clusters | Number of k-means clusters used for conformational clustering. | 5 |
Density axis | Axis used for one-dimensional density profiles. | z |
| Selection string | Meaning |
|---|---|
protein | All atoms in standard amino-acid residues |
protein and name CA | One alpha carbon per residue, often used for coarse-grained structural tracking |
backbone | Backbone atoms N, CA, C, O |
resid 45-70 | Residues 45 through 70 |
protein and not name H* | Heavy atoms only |
resname ATP | All atoms in ATP residues, useful for ligand-focused measurements |
The result page groups outputs into tabs. Not every selected analysis appears in every tab, so the most useful way to read the output is by question rather than by file format.
| Analysis | Output fields | What it answers |
|---|---|---|
RMSD | frame, time_ps, rmsd_angstrom | Is the selected structure drifting away from the reference? |
RMSF | residue_id, residue_name, chain, rmsf_angstrom | Which residues fluctuate the most over the sampled frames? |
Radius of gyration | frame, time_ps, rg_angstrom | Is the selected structure compacting or expanding? |
End-to-end distance | frame, time_ps, distance_angstrom | Are termini or endpoints moving apart or together? |
Asphericity | frame, time_ps, asphericity | Is the shape becoming more elongated or more spherical? |
Moment of inertia | frame, time_ps, I1, I2, I3 | How is mass distribution changing along principal axes? |
| Analysis | Output fields | What it answers |
|---|---|---|
Hydrogen bonds | frame, time_ps, hbond_count; pair table with donor, acceptor, occupancy | Are stabilizing hydrogen-bond networks persistent? |
Salt bridges | frame, time_ps, salt_bridge_count; pair table with residue ids and occupancy | Are charged interactions forming or breaking over time? |
Contacts | frame, time_ps, native_contacts_q, n_contacts | Is an interface retaining its first-frame native contacts? |
Distance tracking | frame, time_ps, distance_angstrom | Is a specific geometric feature moving toward or away from another one? |
H-bond lifetime | lifetime distribution and summary statistics | Are hydrogen bonds brief, recurrent events or long-lived contacts? |
| Analysis | Output fields | What it answers |
|---|---|---|
PCA | frame, time_ps, pc1, pc2, pc3 plus explained variance arrays | What are the dominant collective motions? |
Clustering | per-frame cluster assignments and cluster statistics | How many recurring conformational states are populated? |
MSD | MDAnalysis EinsteinMSD time series and diffusion estimate | Is there translational diffusion or large-scale wandering? |
Dynamic cross-correlation | significant residue pairs and correlation coefficients | Which residue motions are coupled or anticorrelated? |
Persistence length | estimated persistence length and correlation decay | How stiff is a chain-like segment across the sampled trajectory? |
| Analysis | Output fields | What it answers |
|---|---|---|
DSSP | per-frame helix, sheet, and coil percentages | Is secondary structure stable, gained, or lost? |
Ramachandran | residue-level mean and standard deviation for phi and psi | Which residues move into strained or alternative backbone states? |
Chi angles | Janin χ1/χ2 circular means and standard deviations | Are rotamers stable or switching between states? |
Helix analysis | HELANAL rise, twist, bend, tilt, and variability summaries | Are helices remaining canonical or distorting? |
SASA | per-frame total SASA and per-residue SASA summary | Is the structure exposing or burying surface area? |
RDF | radial bins and g(r) values | How is local solvent or particle organization distributed around a selection? |
Density | position bins and density values along one axis | Is mass or solvent redistributed along a membrane or box axis? |
Water dynamics | residence-time summary statistics | How long do hydration waters remain near the selected region? |
RMSD is easiest to misread when alignment and atom selection are not considered together.
Distance tracking is strongest when paired with contacts or hydrogen bonds.
PCA answers whether motion is dominated by a few directions. Clustering answers whether those motions produce recurring states.
These metrics become more informative when read together.
MD trajectory analysis on ProteinIQ is a post-simulation interpretation tool. It is most useful after a trajectory already exists.
| Tool | Best use case | Not the right choice when |
|---|---|---|
MD Trajectory Analysis | Summarizing structure, interaction, and solvent observables from an existing MD run | No trajectory exists yet |
| OpenMM | Running or setting up molecular dynamics simulations | The simulation is already complete and only analysis is needed |
| MDGen | Generating conformational samples when no trajectory exists yet | An experimental or simulated trajectory already exists |
| gmx_MMPBSA | Estimating binding free energy from MD snapshots | The goal is structural interpretation rather than free-energy estimation |
| Ramachandran Plot | Inspecting backbone geometry from a single structure | Time-resolved trajectory behavior is needed |
| RMSD Calculator | Comparing one structure against another | A full time series across many frames is needed |
The main decision point is whether the question is dynamic or static:
protein and name CA is useful for global fold tracking, but it can miss side-chain rearrangements and interaction chemistry.step values reduce runtime and noise, but short-lived events may disappear.
ORB v3 is a universal interatomic potential (machine learning force field) that predicts energies, forces, and stress tensors for atomic systems. Supports both molecular and materials structures with geometry optimization using conservative and direct model variants.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.

Calculate the radius of gyration (Rg) for protein structures from PDB files. Supports multiple chains and atom selection options.
Run molecular dynamics simulations using the GROMACS engine with classical force fields (AMBER, CHARMM, GROMOS, OPLS). Study protein dynamics, conformational flexibility, and structural stability with production-grade MD methodology.
MDGen is a generative AI model for molecular dynamics trajectory generation. Generate physically plausible conformational ensembles from a single protein structure, enabling rapid exploration of protein dynamics without expensive MD simulations.
Run GPU-accelerated molecular dynamics simulations using OpenMM. Simulate protein and protein-ligand complex dynamics with industry-standard force fields (AMBER, CHARMM) and OpenFF ligand parameterization.
Predict molecular energies, atomic forces, atomic and spin charges, dipole vectors, stress tensors, and Hessians from coordinate-bearing molecular structures with AIMNet2 neural network potentials.
Calculate the aliphatic index of protein sequences. A measure of the relative volume occupied by aliphatic side chains, indicating thermostability.
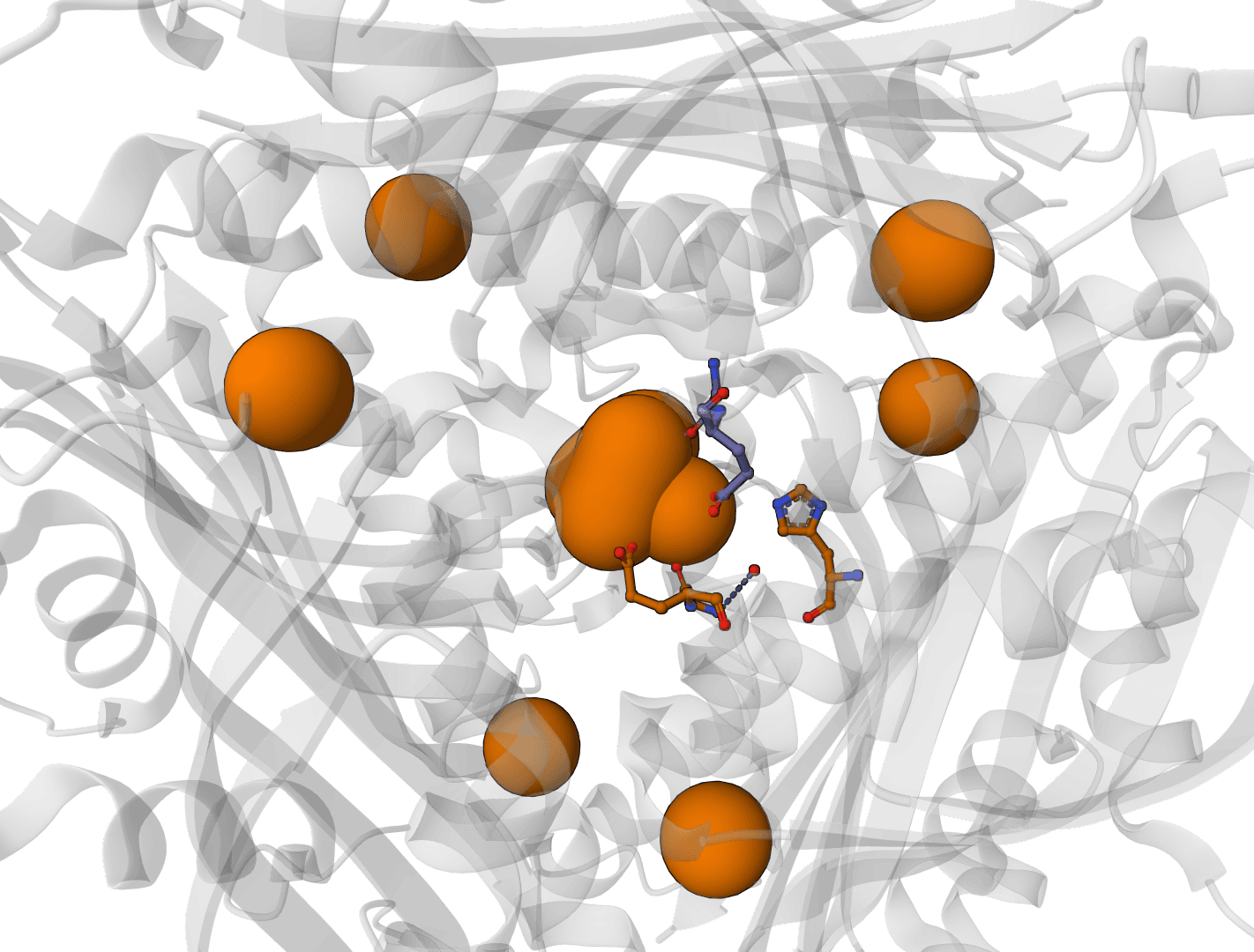
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).
MD trajectory analysis turns a simulation trajectory into measurements that can actually be interpreted: structural drift, residue flexibility, compactness, contact persistence, secondary-structure content, collective motions, and solvent-facing behavior. The raw frames coming out of GROMACS, AMBER, CHARMM, NAMD, or OpenMM do not answer those questions on their own.
This ProteinIQ tool runs a workflow pinned to MDAnalysis 2.9.0. It is not a single command-line program. It is a curated analysis surface built from MDAnalysis modules and a small amount of integration logic that standardizes inputs, runs multiple observables in one job, and returns structured outputs for charting and download.
The practical value is in combining observables that answer different failure modes:
Upload a topology file and a matching trajectory file, choose one or more analyses, and run the job to receive tabular and chart-ready outputs for each selected observable. The result page returns structural, interaction, conformational, environmental, and secondary-structure summaries with downloadable data for follow-up analysis.
| Input | Description | Accepted formats | Max size |
|---|---|---|---|
Topology/Structure file | Defines atom identities, residue assignments, connectivity, and metadata needed to interpret trajectory coordinates. | .pdb, .ent, .gro, .crd, .psf, .prmtop, .top, .tpr | 50 MB |
Trajectory file | Stores coordinates for each sampled frame. The topology and trajectory must describe the same system and atom ordering. | .xtc, .trr, .dcd, .nc, .ncdf, .tng, .crd, .xyz, .mdcrd | 500 MB |
Common pairings include:
.gro + .xtc for GROMACS.tpr + .xtc when topology metadata from GROMACS should be preserved.prmtop + .nc, .ncdf, or .mdcrd for AMBER.psf + .dcd for CHARMM or NAMD.pdb, .crd, or .gro with coordinate trajectories such as .xyz when atom ordering matchesThe tool loads the uploaded files into an MDAnalysis Universe, applies the selected atom selection, resolves the reference mode, optionally aligns sampled frames to that reference, and then dispatches the requested observables.
Three details matter because they change interpretation:
first, last, and average are not cosmetic options. average computes an average reference over the sampled frames instead of aliasing to the first frame.step subsamples the trajectory globally for the audited analysis paths, which reduces runtime and directly changes temporal resolution.Some analyses rely on additional assumptions from the underlying MDAnalysis modules or the tool:
3.0 Å and angle cutoff of 150°, matching the audited MDAnalysis default. Explicit hydrogens are still required for meaningful hydrogen-bond counts.protein and name CA selection is useful for fold tracking but does not contain hydrogens, so lifetime analysis will usually return a warning unless a chemically complete selection is used.MDAnalysis.analysis.contacts.Contacts and reports the source native-contact fraction Q. The displayed contact count is a compatibility summary derived from that fraction and the first-frame native contact set.MDAnalysis.analysis.msd.EinsteinMSD with msd_type="xyz". Physical diffusion estimates require trajectories in an unwrapped coordinate convention.MDAnalysis.analysis.dssp when available. If that path fails, the tool falls back to a simpler internal estimate rather than aborting the whole job.χ1/χ2 summaries use MDAnalysis Janin analysis with circular angle statistics. Helix geometry uses MDAnalysis HELANAL on alpha-carbon selections and requires consecutive helix segments of at least 9 residues.| Setting | Description | Default |
|---|---|---|
Analyses to run | Selects which observables are calculated. Multiple analyses can run in one job. | RMSD, RMSF, Radius of gyration |
Atom selection | MDAnalysis selection string applied to the primary analysis group. Examples include protein, protein and name CA, backbone, and resid 1-100. | protein and name CA |
Reference frame | Reference for RMSD-aligned analyses. Average structure computes the mean coordinate set over the sampled frames. | first |
| Setting | Description | Default |
|---|---|---|
Frame step | Samples every nth frame. A value of 10 processes frames 0, 10, 20, ... instead of every saved frame. | 1 |
Align trajectory | Aligns sampled frames to the chosen reference before RMSD, RMSF, PCA, clustering, and cross-correlation analysis. | true |
Second atom selection | Secondary MDAnalysis selection used for distance tracking and RDF. | resname SOL and name OW |
PCA components | Number of principal components returned in PCA projections. | 3 |
Clusters | Number of k-means clusters used for conformational clustering. | 5 |
Density axis | Axis used for one-dimensional density profiles. | z |
| Selection string | Meaning |
|---|---|
protein | All atoms in standard amino-acid residues |
protein and name CA | One alpha carbon per residue, often used for coarse-grained structural tracking |
backbone | Backbone atoms N, CA, C, O |
resid 45-70 | Residues 45 through 70 |
protein and not name H* | Heavy atoms only |
resname ATP | All atoms in ATP residues, useful for ligand-focused measurements |
The result page groups outputs into tabs. Not every selected analysis appears in every tab, so the most useful way to read the output is by question rather than by file format.
| Analysis | Output fields | What it answers |
|---|---|---|
RMSD | frame, time_ps, rmsd_angstrom | Is the selected structure drifting away from the reference? |
RMSF | residue_id, residue_name, chain, rmsf_angstrom | Which residues fluctuate the most over the sampled frames? |
Radius of gyration | frame, time_ps, rg_angstrom | Is the selected structure compacting or expanding? |
End-to-end distance | frame, time_ps, distance_angstrom | Are termini or endpoints moving apart or together? |
Asphericity | frame, time_ps, asphericity | Is the shape becoming more elongated or more spherical? |
Moment of inertia | frame, time_ps, I1, I2, I3 | How is mass distribution changing along principal axes? |
| Analysis | Output fields | What it answers |
|---|---|---|
Hydrogen bonds | frame, time_ps, hbond_count; pair table with donor, acceptor, occupancy | Are stabilizing hydrogen-bond networks persistent? |
Salt bridges | frame, time_ps, salt_bridge_count; pair table with residue ids and occupancy | Are charged interactions forming or breaking over time? |
Contacts | frame, time_ps, native_contacts_q, n_contacts | Is an interface retaining its first-frame native contacts? |
Distance tracking | frame, time_ps, distance_angstrom | Is a specific geometric feature moving toward or away from another one? |
H-bond lifetime | lifetime distribution and summary statistics | Are hydrogen bonds brief, recurrent events or long-lived contacts? |
| Analysis | Output fields | What it answers |
|---|---|---|
PCA | frame, time_ps, pc1, pc2, pc3 plus explained variance arrays | What are the dominant collective motions? |
Clustering | per-frame cluster assignments and cluster statistics | How many recurring conformational states are populated? |
MSD | MDAnalysis EinsteinMSD time series and diffusion estimate | Is there translational diffusion or large-scale wandering? |
Dynamic cross-correlation | significant residue pairs and correlation coefficients | Which residue motions are coupled or anticorrelated? |
Persistence length | estimated persistence length and correlation decay | How stiff is a chain-like segment across the sampled trajectory? |
| Analysis | Output fields | What it answers |
|---|---|---|
DSSP | per-frame helix, sheet, and coil percentages | Is secondary structure stable, gained, or lost? |
Ramachandran | residue-level mean and standard deviation for phi and psi | Which residues move into strained or alternative backbone states? |
Chi angles | Janin χ1/χ2 circular means and standard deviations | Are rotamers stable or switching between states? |
Helix analysis | HELANAL rise, twist, bend, tilt, and variability summaries | Are helices remaining canonical or distorting? |
SASA | per-frame total SASA and per-residue SASA summary | Is the structure exposing or burying surface area? |
RDF | radial bins and g(r) values | How is local solvent or particle organization distributed around a selection? |
Density | position bins and density values along one axis | Is mass or solvent redistributed along a membrane or box axis? |
Water dynamics | residence-time summary statistics | How long do hydration waters remain near the selected region? |
RMSD is easiest to misread when alignment and atom selection are not considered together.
Distance tracking is strongest when paired with contacts or hydrogen bonds.
PCA answers whether motion is dominated by a few directions. Clustering answers whether those motions produce recurring states.
These metrics become more informative when read together.
MD trajectory analysis on ProteinIQ is a post-simulation interpretation tool. It is most useful after a trajectory already exists.
| Tool | Best use case | Not the right choice when |
|---|---|---|
MD Trajectory Analysis | Summarizing structure, interaction, and solvent observables from an existing MD run | No trajectory exists yet |
| OpenMM | Running or setting up molecular dynamics simulations | The simulation is already complete and only analysis is needed |
| MDGen | Generating conformational samples when no trajectory exists yet | An experimental or simulated trajectory already exists |
| gmx_MMPBSA | Estimating binding free energy from MD snapshots | The goal is structural interpretation rather than free-energy estimation |
| Ramachandran Plot | Inspecting backbone geometry from a single structure | Time-resolved trajectory behavior is needed |
| RMSD Calculator | Comparing one structure against another | A full time series across many frames is needed |
The main decision point is whether the question is dynamic or static:
protein and name CA is useful for global fold tracking, but it can miss side-chain rearrangements and interaction chemistry.step values reduce runtime and noise, but short-lived events may disappear.ORB v3 is a universal interatomic potential (machine learning force field) that predicts energies, forces, and stress tensors for atomic systems. Supports both molecular and materials structures with geometry optimization using conservative and direct model variants.
Faithful static-mode Aggrescan3D tool for per-residue aggregation propensity analysis from a single protein structure.
Predict pKa values of ionizable groups in proteins and protein-ligand complexes from 3D structure. PROPKA calculates environment-driven pKa shifts for standard ionizable residues, terminal groups, and supported ligand atom types.
Calculate the radius of gyration (Rg) for protein structures from PDB files. Supports multiple chains and atom selection options.
Run molecular dynamics simulations using the GROMACS engine with classical force fields (AMBER, CHARMM, GROMOS, OPLS). Study protein dynamics, conformational flexibility, and structural stability with production-grade MD methodology.
MDGen is a generative AI model for molecular dynamics trajectory generation. Generate physically plausible conformational ensembles from a single protein structure, enabling rapid exploration of protein dynamics without expensive MD simulations.
Run GPU-accelerated molecular dynamics simulations using OpenMM. Simulate protein and protein-ligand complex dynamics with industry-standard force fields (AMBER, CHARMM) and OpenFF ligand parameterization.
Predict molecular energies, atomic forces, atomic and spin charges, dipole vectors, stress tensors, and Hessians from coordinate-bearing molecular structures with AIMNet2 neural network potentials.
Calculate the aliphatic index of protein sequences. A measure of the relative volume occupied by aliphatic side chains, indicating thermostability.
Predict metal and water binding sites in protein structures using 3D convolutional neural networks (AllMetal3D + Water3D).