Design antibody CDR loops from antibody-antigen complex structures using inverse folding. Learn more
Design antibody CDR loops from antibody-antigen complex structures using inverse folding. Learn more
Design antibody CDR loops from antibody-antigen complex structures using inverse folding. Learn more
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
Design thermostable protein sequences using ProteinMPNN trained on hyperthermophilic organism structures. Generates sequences optimized for improved thermal stability without requiring ligands or additional context.
Design protein sequences with atomic context from ligands, metals, and nucleotides. Achieves 63.3% sequence recovery at binding sites, significantly outperforming ProteinMPNN (50.5%).
Design protein sequences for given backbone structures using deep learning. Fast and accurate inverse folding with state-of-the-art sequence recovery (52.4%).
Specialized model for soluble protein sequence design. Trained exclusively on soluble proteins for optimized performance on cytoplasmic and extracellular proteins.
Humatch is an antibody humanization tool that transforms non-human antibody sequences into humanized variants. Uses three lightweight CNNs to identify optimal human V-genes and generate paired heavy and light chain sequences with minimal edits while maintaining functionality.
Design VHH nanobody binders using AlphaFold-Multimer with structure templates and sequence conditioning. mBER (Manifold Binder Engineering and Refinement) generates novel VHH antibody sequences that bind to user-specified target proteins.
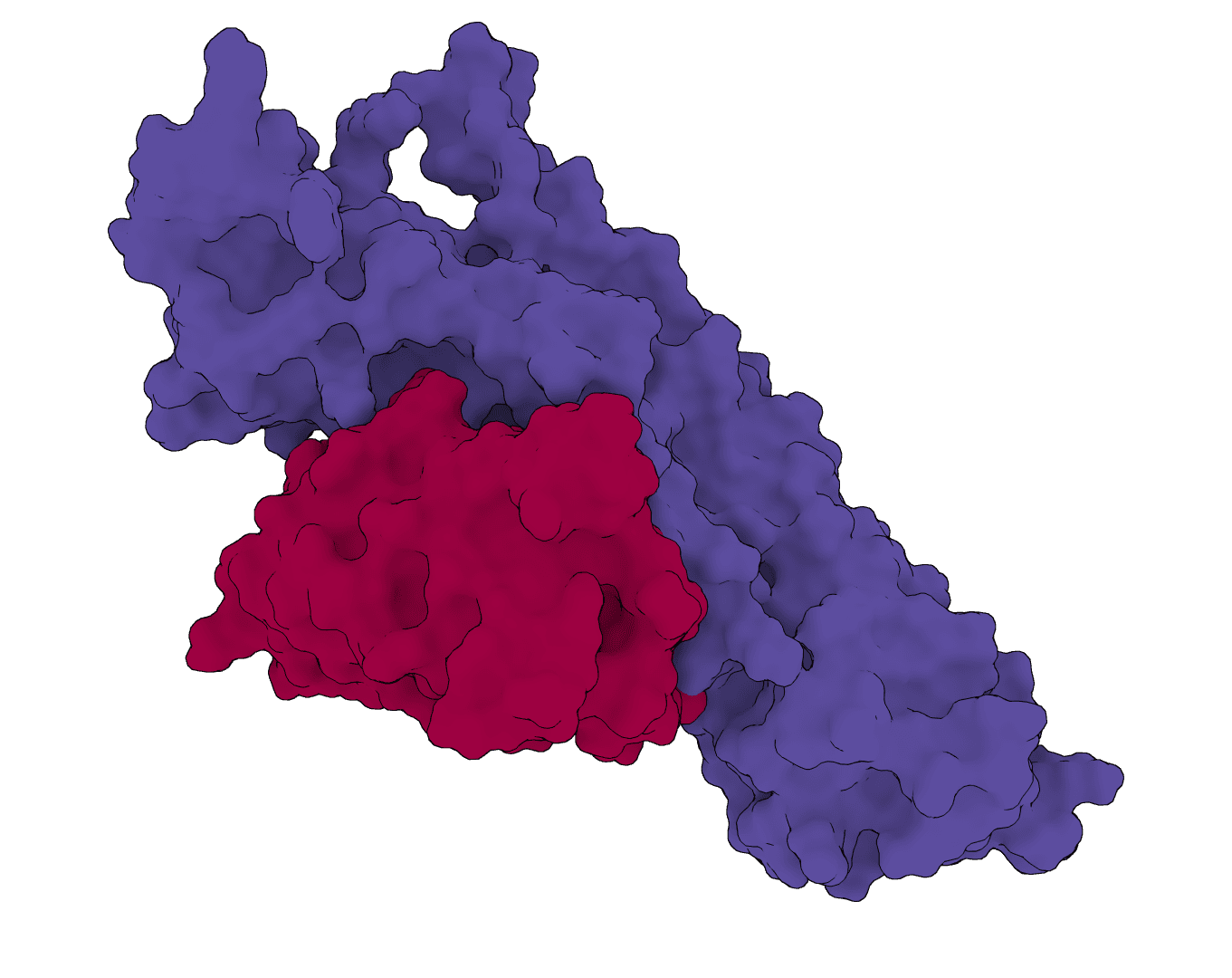
Structure-based de novo antibody and nanobody design pipeline combining antibody-tuned RFdiffusion, ProteinMPNN sequence design, and antibody-tuned RoseTTAFold2 filtering.
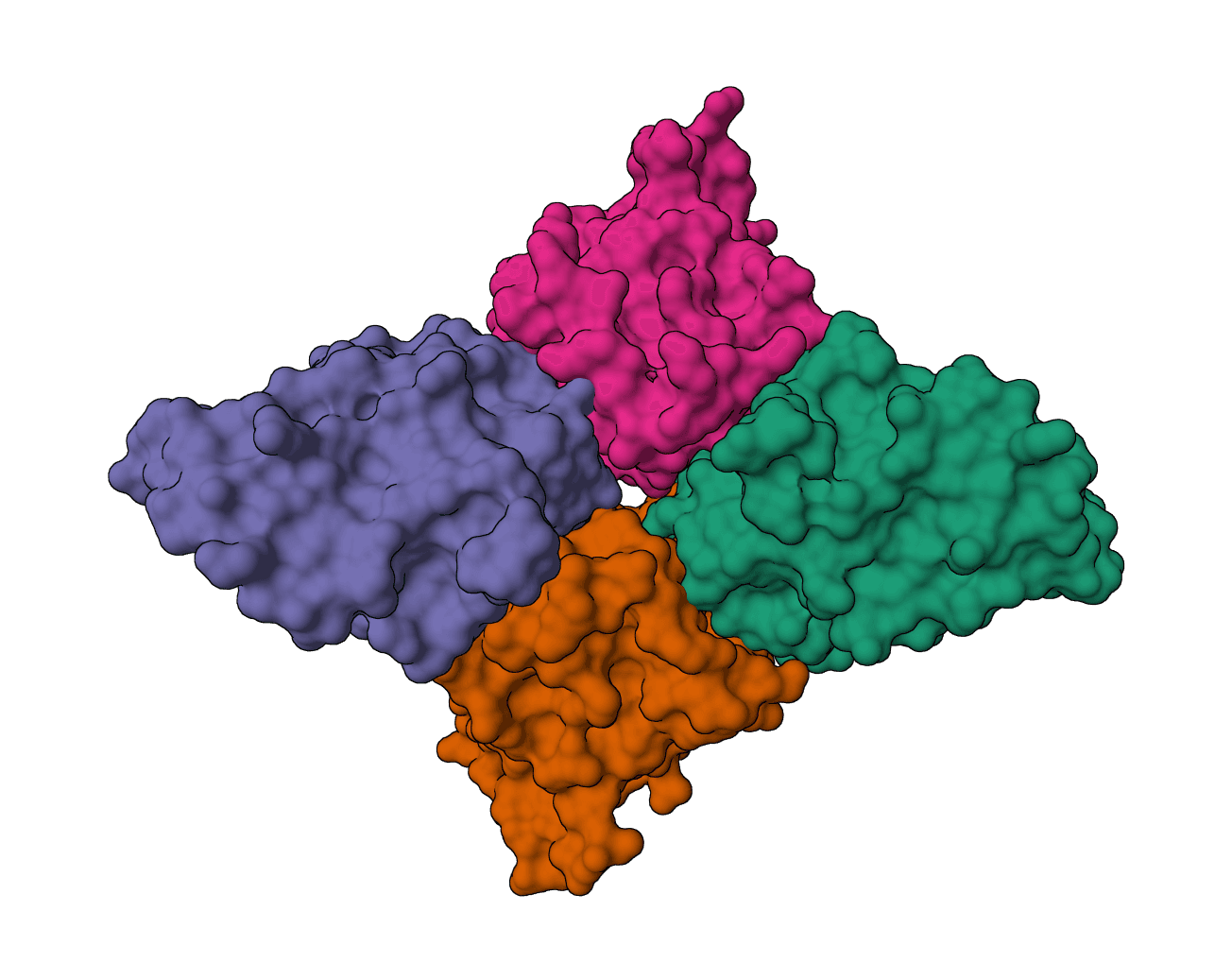
Antibody humanization and humanness evaluation platform from Merck. Sapiens mode uses deep learning trained on the Observed Antibody Space (OAS) to humanize antibody sequences, while OASis mode evaluates humanness using 9-mer peptide search against human antibody databases.
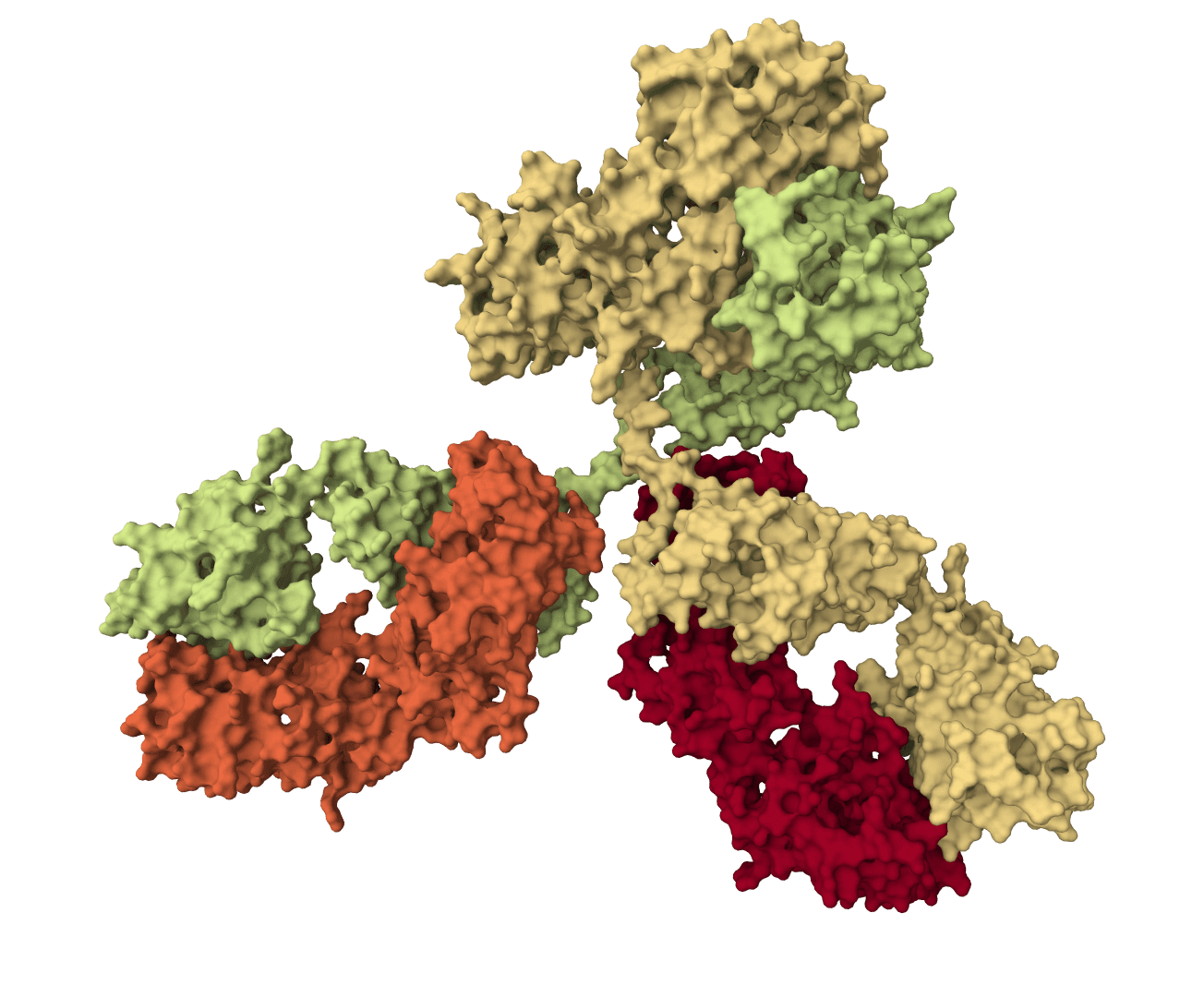
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
Design thermostable protein sequences using ProteinMPNN trained on hyperthermophilic organism structures. Generates sequences optimized for improved thermal stability without requiring ligands or additional context.
Design protein sequences with atomic context from ligands, metals, and nucleotides. Achieves 63.3% sequence recovery at binding sites, significantly outperforming ProteinMPNN (50.5%).
Design protein sequences for given backbone structures using deep learning. Fast and accurate inverse folding with state-of-the-art sequence recovery (52.4%).
Specialized model for soluble protein sequence design. Trained exclusively on soluble proteins for optimized performance on cytoplasmic and extracellular proteins.
Humatch is an antibody humanization tool that transforms non-human antibody sequences into humanized variants. Uses three lightweight CNNs to identify optimal human V-genes and generate paired heavy and light chain sequences with minimal edits while maintaining functionality.
Design VHH nanobody binders using AlphaFold-Multimer with structure templates and sequence conditioning. mBER (Manifold Binder Engineering and Refinement) generates novel VHH antibody sequences that bind to user-specified target proteins.
Structure-based de novo antibody and nanobody design pipeline combining antibody-tuned RFdiffusion, ProteinMPNN sequence design, and antibody-tuned RoseTTAFold2 filtering.
Antibody humanization and humanness evaluation platform from Merck. Sapiens mode uses deep learning trained on the Observed Antibody Space (OAS) to humanize antibody sequences, while OASis mode evaluates humanness using 9-mer peptide search against human antibody databases.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
Configure inputs to begin
Set options on the left, then click “Submit job”.
IgDesign is an inverse-folding model from Absci for designing antibody complementarity-determining region (CDR) sequences from an antibody-antigen complex structure. It conditions sequence generation on the antibody backbone, framework sequence, light chain, and antigen context.
IgDesign can redesign heavy-chain CDRs (HCDR1–3), light-chain CDRs (LCDR1–3), or a selected combination. ProteinIQ runs the reviewed IgDesign 1.0.0 source revision on a cloud GPU and preserves its native prediction table.
Submit exactly one PDB structure or RCSB PDB ID. The structure must contain:
ATOM records for all three selected chain IDs; andSEQRES records for all three selected chains.IgDesign’s parser compares the coordinate-derived sequence with SEQRES. Structures without matching records are rejected before the model runs.
Each chain ID must be one alphanumeric PDB chain character. The defaults are H for heavy, L for light, and A for antigen.
Select one or more heavy- or light-chain CDRs. ProteinIQ uses IgDesign’s own PDB parser and ANARCI integration to annotate each antibody chain in the IMGT scheme, then passes the resulting sequence indices to IgDesign. Positions are therefore derived from the submitted structure rather than assumed from a fixed example.
IgDesign’s native output count is:
decoding orders × ProteinMPNN samples × language-model samples
All three settings default to 10, producing 1,000 rows. Reducing any factor reduces both output count and compute. The ProteinMPNN and language-model temperatures independently control diversity at their corresponding sampling stages; both default to 0.5.
The random seed defaults to 0, matching the reviewed source configuration. Independent CDR loss calculation is enabled by default.
The Results tab displays every column from IgDesign’s native CSV in source order. Selected CDR columns contain generated sequences. When independent loss is enabled, the source adds CDR-specific columns such as ce_loss_independent_hcdr1; each cell contains pipe-delimited per-position loss values.
The Files tab includes:
Cross-entropy loss is a model score for the generated sequence under the configured structural context. IgDesign does not define universal score bands or claim that a particular loss guarantees binding affinity, so compare candidates within the same run and validate selected designs experimentally.
Shanehsazzadeh A, Alverio J, Kasun G, Levine S, et al. IgDesign: Integrated Generative Modeling for Antibody Design. bioRxiv (2023). doi:10.1101/2023.12.08.570889