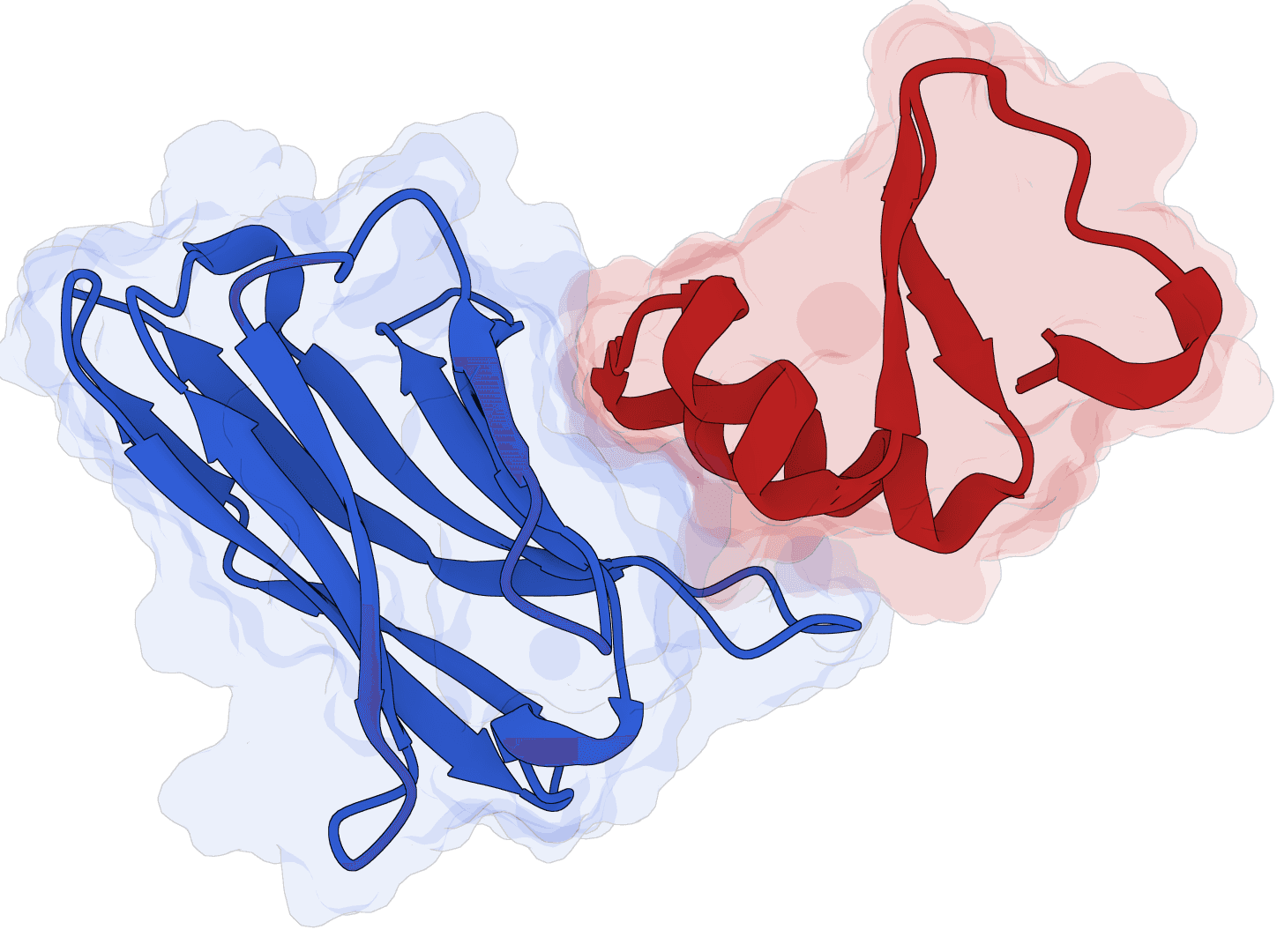
Structure-based de novo antibody and nanobody design pipeline combining antibody-tuned RFdiffusion, ProteinMPNN sequence design, and antibody-tuned RoseTTAFold2 filtering.
Humatch is an antibody humanization tool that transforms non-human antibody sequences into humanized variants. Uses three lightweight CNNs to identify optimal human V-genes and generate paired heavy and light chain sequences with minimal edits while maintaining functionality.
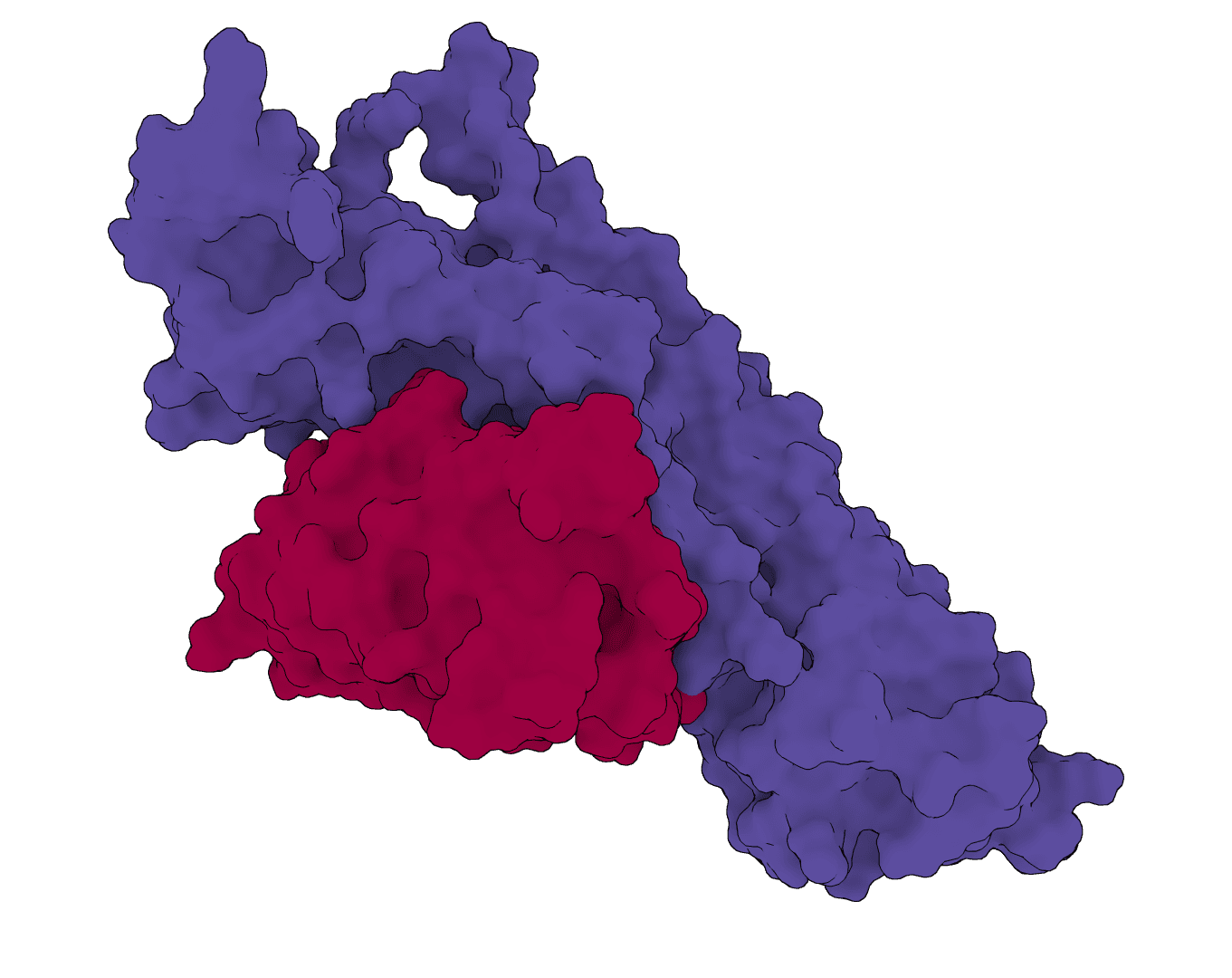
Design antibody heavy- and light-chain CDR sequences from an antibody-antigen complex with the IgDesign inverse-folding model.
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
Antibody humanization and humanness evaluation platform from Merck. Sapiens mode uses deep learning trained on the Observed Antibody Space (OAS) to humanize antibody sequences, while OASis mode evaluates humanness using 9-mer peptide search against human antibody databases.
IgGM is a generative foundation model for antibody and nanobody design against a target antigen. Supports CDR design, affinity maturation, inverse design, and framework design. Requires an antigen structure (PDB) and antibody sequences with "X" marking positions to design.
Design de novo protein binders using AlphaFold2 backpropagation, ProteinMPNN sequence optimization, and PyRosetta relaxation. BindCraft generates novel protein sequences that bind to user-specified target surfaces.
Optimize protein binders using genetic algorithms combined with AlphaFold2 fitness evaluation and ProteinMPNN sequence design. EvoPro evolves protein sequences to maximize binding affinity and structural quality through iterative cycles of mutation, selection, and validation.
Structure-based de novo antibody and nanobody design pipeline combining antibody-tuned RFdiffusion, ProteinMPNN sequence design, and antibody-tuned RoseTTAFold2 filtering.
Humatch is an antibody humanization tool that transforms non-human antibody sequences into humanized variants. Uses three lightweight CNNs to identify optimal human V-genes and generate paired heavy and light chain sequences with minimal edits while maintaining functionality.
Design antibody heavy- and light-chain CDR sequences from an antibody-antigen complex with the IgDesign inverse-folding model.
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
BoltzGen uses generative diffusion models to design protein, peptide, nanobody, and Fab binders against protein and small-molecule targets.
Exploratory antibody CDR co-design for antibody-antigen complexes using Proteo-R1 reasoning and raw diffusion. The standard online workflow does not include the framework structure-inpainting assets required for the published-quality target.
Antibody humanization and humanness evaluation platform from Merck. Sapiens mode uses deep learning trained on the Observed Antibody Space (OAS) to humanize antibody sequences, while OASis mode evaluates humanness using 9-mer peptide search against human antibody databases.
IgGM is a generative foundation model for antibody and nanobody design against a target antigen. Supports CDR design, affinity maturation, inverse design, and framework design. Requires an antigen structure (PDB) and antibody sequences with "X" marking positions to design.
Design de novo protein binders using AlphaFold2 backpropagation, ProteinMPNN sequence optimization, and PyRosetta relaxation. BindCraft generates novel protein sequences that bind to user-specified target surfaces.
Optimize protein binders using genetic algorithms combined with AlphaFold2 fitness evaluation and ProteinMPNN sequence design. EvoPro evolves protein sequences to maximize binding affinity and structural quality through iterative cycles of mutation, selection, and validation.
Configure inputs to begin
Set options on the left, then click “Submit job” — or start from an example.
Ubiquitin — 5-minute budget exhausted before first trajectory
PD-L1 A56 — accepted VHH design (iPTM 0.759)
mBER (Manifold Binder Engineering and Refinement) designs de novo VHH nanobody sequences against a chosen protein surface. It combines a VHH structural template, antibody sequence conditioning, and AlphaFold-Multimer backpropagation so the search stays within an antibody format while optimizing a predicted binder-target complex.
The 2025 mBER preprint reports more than one million designs across 436 targets, with experimental screening against 145 targets. The authors reported specific, statistically significant design success against 45% of screened targets and binding rates up to 38% for selected epitopes after filtering. The reported performance belongs to that screening campaign and does not establish an expected rate for a new target or individual design.
Run mBER online by uploading a target PDB structure or fetching one from RCSB. Select the target chains, optionally name hotspot residues, and set the search and acceptance criteria. ProteinIQ returns accepted VHH sequences, predicted complexes, confidence metrics, downloadable files, and, when requested, evaluated candidates that fell below the thresholds.
| Input | Description |
|---|---|
Target Protein | One protein structure in .pdb or .ent format, up to 50 MB. A structure can also be fetched from RCSB with a four-character PDB ID such as 5JDS. Include the chains needed to represent the intended target surface. |
When no experimental structure is available, a predicted structure from AlphaFold2 can be downloaded as PDB and uploaded. Low-confidence loops, unresolved residues, alternate conformations, and flexible domains can make the designed interface less reliable.
| Setting | Description |
|---|---|
Target chains | Chain ID or comma-separated chain list to design against, for example A or A,B. The default is A. The IDs must match the submitted PDB. |
Hotspot residues (optional) | Comma-separated target residues such as A56,A60 or A56,B20. Hotspots steer the search toward a surface patch. Leaving the field empty allows mBER to search without a specified epitope. |
Number of designs | Number of accepted VHH designs requested, from 1 to 500. ProteinIQ defaults to 1. More designs usually require more successful trajectories and a longer search. |
Maximum trajectories | Maximum number of independent design attempts, from 1 to 10000 (default 10000). The run can stop earlier after finding the requested number of accepted designs or reaching its runtime limit. |
Minimum iPTM | Acceptance threshold for interface predicted TM score, from 0.50 to 0.95 (default 0.75). Raising it makes the interface filter stricter. |
Minimum pLDDT | Acceptance threshold for normalized pLDDT, from 0.50 to 0.95 (default 0.70). Raising it makes the local-structure filter stricter. |
Show below-threshold candidates | Includes evaluated candidates that failed one or both acceptance thresholds. Enabled by default. These rows are exploratory results, not accepted mBER designs. |
Target name (optional) | Name passed to mBER and used in run identifiers. When empty, the name comes from the uploaded filename or RCSB identifier. |
The iPTM and pLDDT thresholds are filters on model confidence. They do not set a binding-affinity cutoff, and lowering them does not improve a candidate. It only keeps more uncertain candidates in the accepted set.
The Viewer tab loads the preferred PDB for each candidate. A relaxed structure is preferred when available; otherwise, the predicted complex is shown. The Data tab keeps accepted designs in native acceptance order and can also include below-threshold candidates. The Files tab contains the submitted target, returned PDBs, accepted.csv, and retained artifacts for accepted trajectories.
| Result | Description |
|---|---|
Status | accepted when the candidate passed the configured iPTM and pLDDT filters, or below_threshold when it was returned for exploration. |
Trajectory | Name of the independent mBER design attempt that produced the candidate. |
Binder | Binder index within the trajectory. |
Sequence | Designed VHH amino acid sequence. |
iPTM | Confidence in the predicted relative placement of binder and target chains, on a 0 to 1 scale. Higher is better. |
pLDDT | Local structural confidence reported by mBER on a normalized 0 to 1 scale. Multiply by 100 to compare with the familiar AlphaFold pLDDT scale. |
pTM | Confidence in the overall fold and arrangement of the predicted complex, on a 0 to 1 scale. |
PAE | Predicted aligned error summary when present. Lower values indicate greater confidence in relative geometry. |
Interface PAE | PAE summary focused on the binder-target interface when present. Lower is better. |
Seq Entropy | Native sequence-entropy value retained from trajectory evaluation. Use it for comparison within the same run rather than as a universal acceptance threshold. |
ESM Score | Native language-model score when present. Its value is useful for relative comparison among candidates produced under the same settings. |
Relaxed Energy | Energy reported after structural relaxation when available. Compare values only among structures prepared by the same run and protocol. |
Complex PDB | Predicted binder-target complex before relaxation. |
Relaxed PDB | Relaxed binder-target complex when relaxation completed. |
Monomer PDB | Binder-only structure retained from trajectory evaluation when available. |
accepted.csv contains the sequence and core metrics for candidates that passed the filters. Below-threshold candidates come from retained trajectory evaluation data and do not become accepted rows in that file.
Start with Status. An accepted candidate passed the chosen computational filters; it has not been experimentally confirmed. When Show below-threshold candidates is enabled, the table may contain structurally interesting candidates even if the run produced no accepted designs.
Next, read the confidence metrics together:
0.8 indicate a confident complex prediction, values from 0.6 to 0.8 are uncertain, and values below 0.6 often indicate a failed complex prediction. These ranges are model-confidence guidance, not probabilities of binding.Visual inspection still matters. The binder should contact the intended surface without severe clashes, disconnected geometry, or an interface dominated by unresolved target regions. With hotspots, check that the modeled contacts actually involve the requested residues. Hotspots bias optimization but do not guarantee that every final pose centers on them.
A completed run with zero accepted designs is a valid search outcome. The target may be difficult, the requested epitope may be inaccessible, the thresholds may be strict, or the search may have ended at the trajectory or runtime limit.
Below-threshold rows show whether the search approached the acceptance boundary. Candidates with iPTM or pLDDT just under the threshold may justify a broader exploratory run. Uniformly weak interfaces call for a different target structure, revised chain selection, or a more accessible hotspot rather than a small threshold change.
This completed public job shows the structure, confidence metrics, and downloadable artifacts returned by mBER. Open the example to inspect the interactive result and reuse its inputs and settings.
This example targets chain A of a PD-L1 structure and directs the VHH search toward residue A56. It requests one accepted design from one trajectory, making it a compact example of a successful targeted run.
PDL1.pdb, with Target chains = AHotspot residues (optional) = A56; Maximum trajectories = 1; Minimum iPTM = 0.50; Minimum pLDDT = 0.50; Target name (optional) = PDL1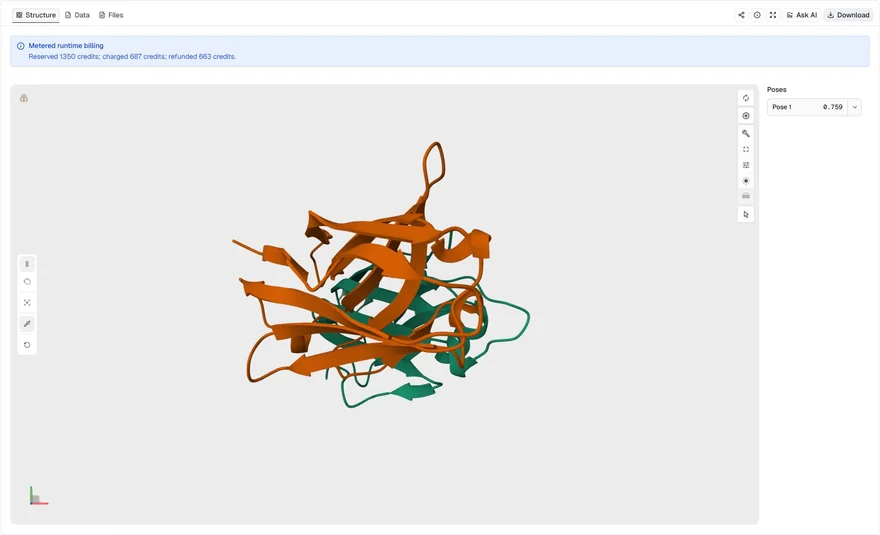
mBER accepted one VHH sequence from trajectory PDL1_1212235. The accepted row reports iPTM 0.7588, pLDDT 0.9276, pTM 0.8341, PAE 0.1531, and Interface PAE 0.1452. The viewer selects the relaxed complex PDB when available, so the displayed structure is the run's preferred model for inspecting the proposed binder-target geometry.
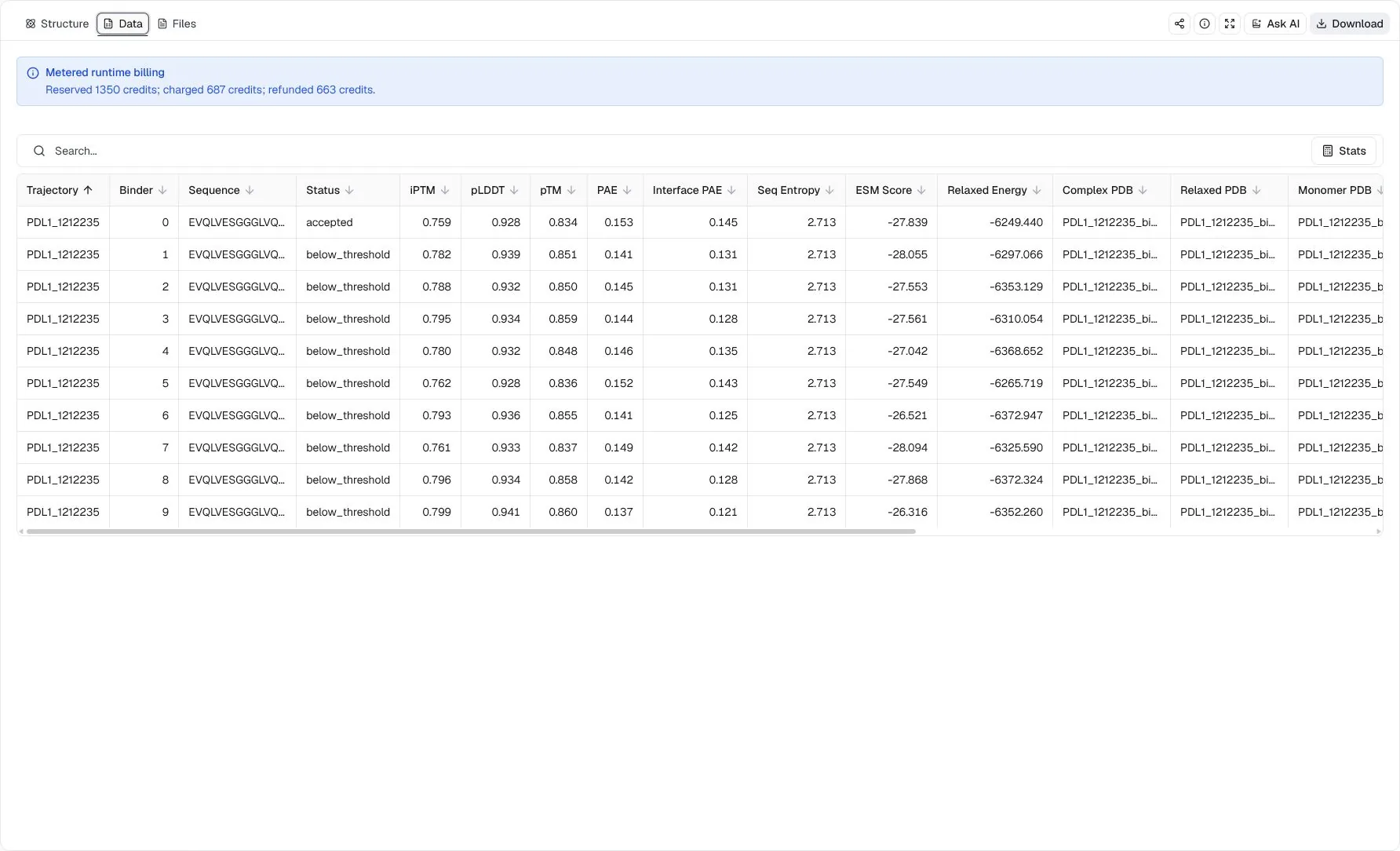
The Data view also includes nine retained evaluation candidates because Show below-threshold candidates was enabled. Some have higher individual confidence scores than the accepted row, but they are still labeled below_threshold rather than included in native accepted.csv. Treat them as exploratory alternatives, not as accepted designs or a score-ranked replacement for the accepted sequence.
The job completed in 15 minutes 15 seconds. These confidence metrics and the modeled interface support structural review and candidate prioritization; they do not demonstrate binding, affinity, specificity, or experimental activity.
mBER prepares the selected target chains and builds a VHH starting structure with NanoBodyBuilder2-derived structural information. The antibody framework constrains the search to a single-domain antibody geometry instead of an unconstrained mini-protein scaffold.
Optional hotspots define residues that should contribute to the proposed interface. Without hotspots, the method can explore the accessible target surface more broadly.
The method adds antibody sequence priors from protein language models to the VHH template. These priors favor sequence patterns that resemble the model's learned antibody distribution while leaving design positions free to adapt to the target. The published method combines this sequence information with structural conditioning and does not train a new folding or language model.
Each trajectory optimizes a VHH sequence against AlphaFold-Multimer predictions of the binder-target complex. ColabDesign-style backpropagation updates the sequence to improve structural and interface objectives. Independent trajectories start from different search states, so repeating or expanding a run can produce different candidates.
mBER evaluates each trajectory and keeps candidates that meet both Minimum iPTM and Minimum pLDDT. Accepted candidates are written to accepted.csv with their structures and metrics. ProteinIQ can also recover evaluated candidates below those thresholds, making a failed acceptance search easier to diagnose without labeling the candidates as successful designs.
| Method | Best fit |
|---|---|
| mBER | Designing new VHH nanobody sequences against a known target structure, with optional epitope control. |
| BindCraft | Designing compact de novo protein binders when an antibody scaffold is not required. |
| DiffAb | Redesigning one or more CDR loops in an existing antibody-antigen complex. |
| AlphaFold2 | Predicting the structure of an existing sequence or complex rather than generating a new binder sequence. |
mBER is VHH-specific. It does not design paired heavy and light chains, predict an equilibrium dissociation constant, or replace developability assessment. A high-confidence complex can still fail because of expression, aggregation, off-target binding, epitope occlusion, or inaccurate target conformations. Experimental expression, specificity, and affinity measurements remain necessary before advancing a design.