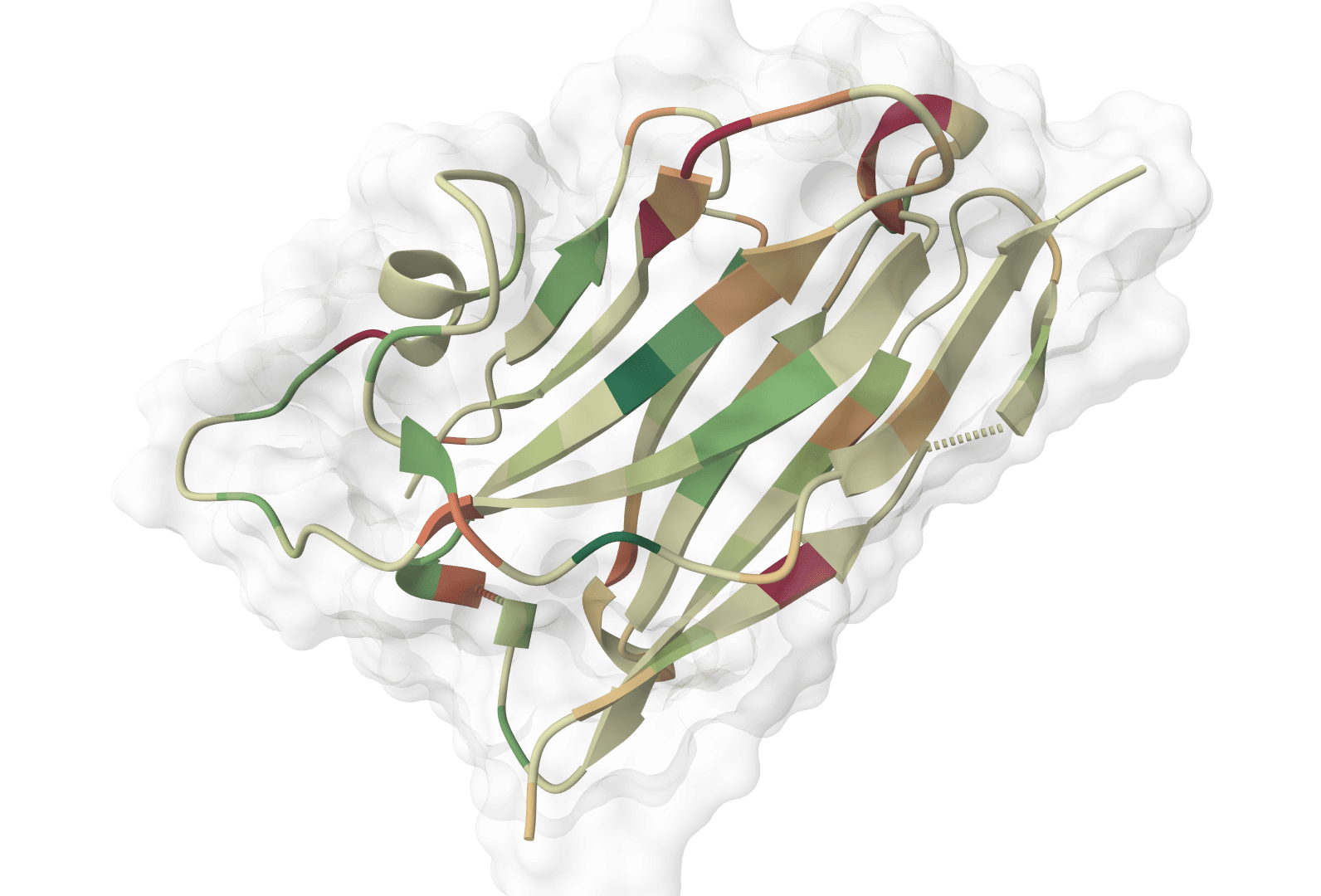
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
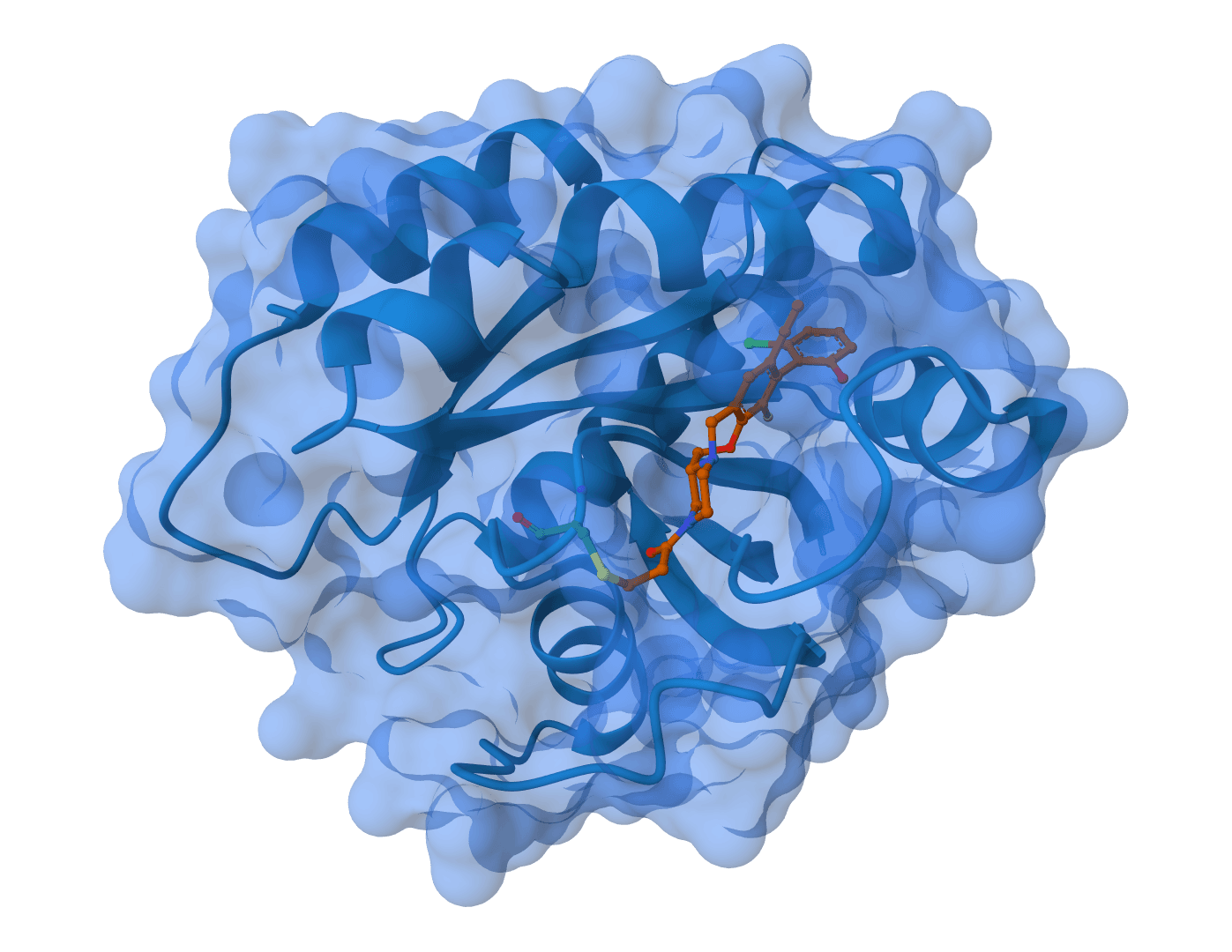
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
Controllable all-atom structure prediction for proteins, ligands, DNA, RNA, and multi-component complexes using IntelliFold 2.0.4 on its AlphaFold 3 JAX engine.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
MiniFold is a fast single-sequence protein structure predictor that is 10-20x faster than ESMFold. It predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it ideal for rapid structure prediction.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
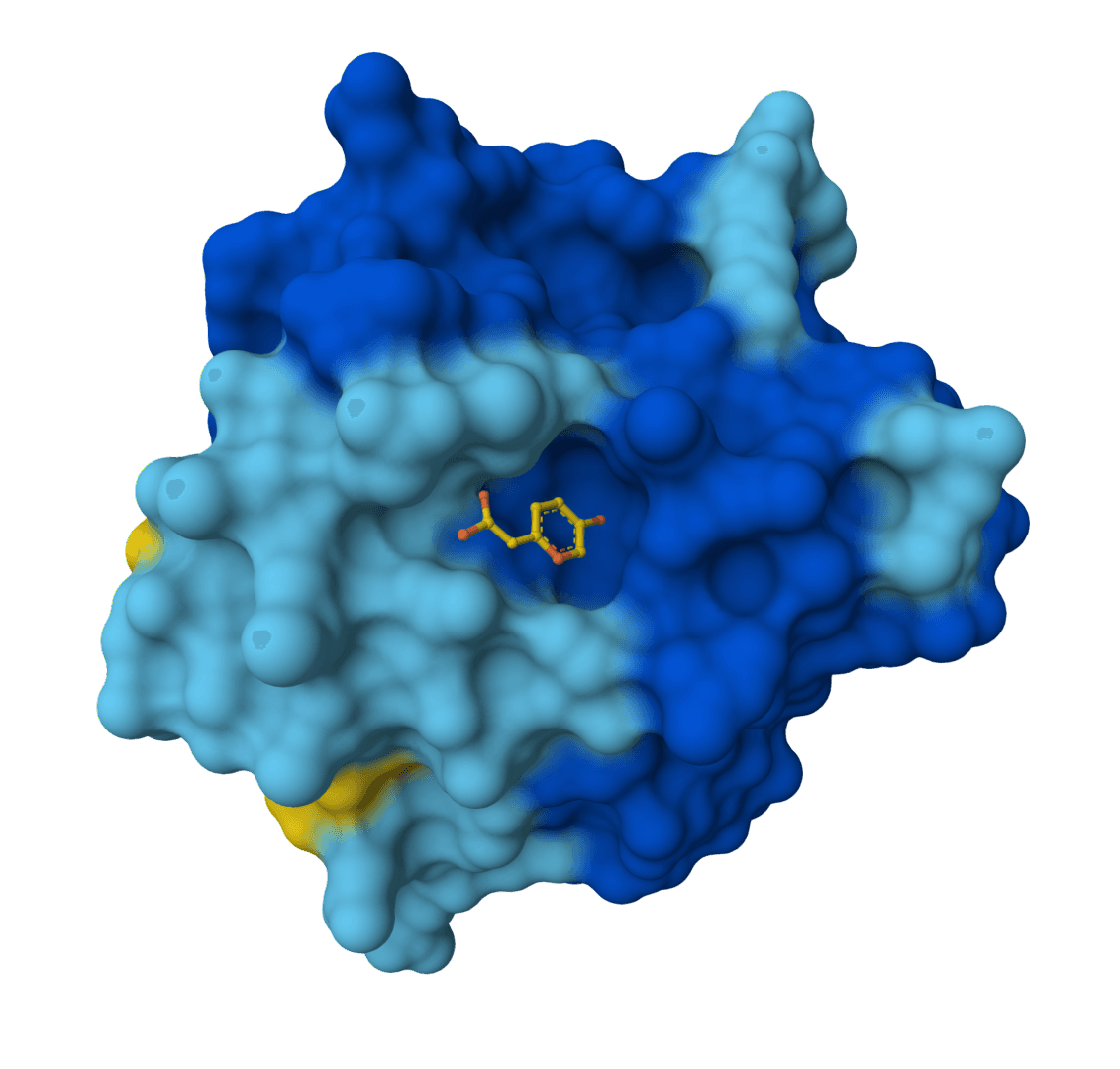
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
Controllable all-atom structure prediction for proteins, ligands, DNA, RNA, and multi-component complexes using IntelliFold 2.0.4 on its AlphaFold 3 JAX engine.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
MiniFold is a fast single-sequence protein structure predictor that is 10-20x faster than ESMFold. It predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it ideal for rapid structure prediction.
Configure inputs to begin
Set options on the left, then click “Submit job”.
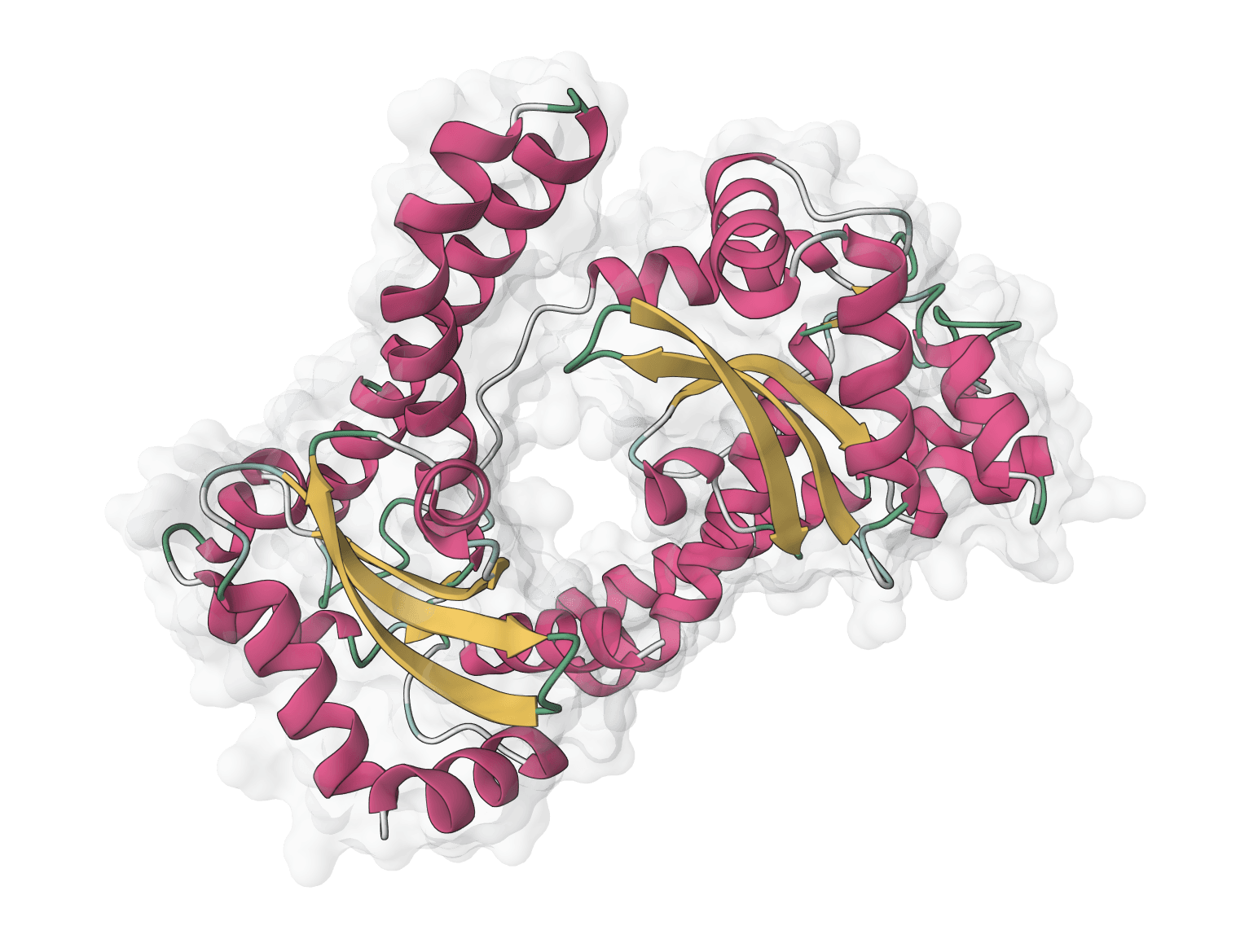
ImmuneBuilder is a suite of deep learning models for predicting 3D structures of immune receptor proteins. Developed at the Oxford Protein Informatics Group and published in Communications Biology in 2023, it includes three specialized predictors: ABodyBuilder3 for antibodies, NanoBodyBuilder2 for nanobodies, and TCRBuilder2 for T-cell receptors.
The key advantage over general-purpose structure predictors like AlphaFold2 is speed. By training exclusively on immune receptor structures, ImmuneBuilder achieves over 100x faster predictions while matching or exceeding AlphaFold2's accuracy for CDR loop conformations. This makes it practical for high-throughput screening of antibody libraries and therapeutic antibody engineering workflows where hundreds or thousands of structures need rapid evaluation.
On benchmarks of recently solved antibody structures, ABodyBuilder2 predicts CDR-H3 loops with an RMSD of 2.81 Angstroms—a 0.09 Angstrom improvement over AlphaFold-Multimer. NanoBodyBuilder2 achieves 2.89 Angstrom RMSD on CDR-H3 loops, outperforming AlphaFold2 by 0.55 Angstroms.
ImmuneBuilder adapts the structure module architecture from AlphaFold-Multimer with modifications optimized for antibody-like proteins. Each residue is represented as a rigid body defined by 3D coordinates and orientation matrices. Eight sequential update blocks process these representations using Invariant Point Attention layers for structurally-aware feature updates and Backbone Update layers for coordinate refinement.
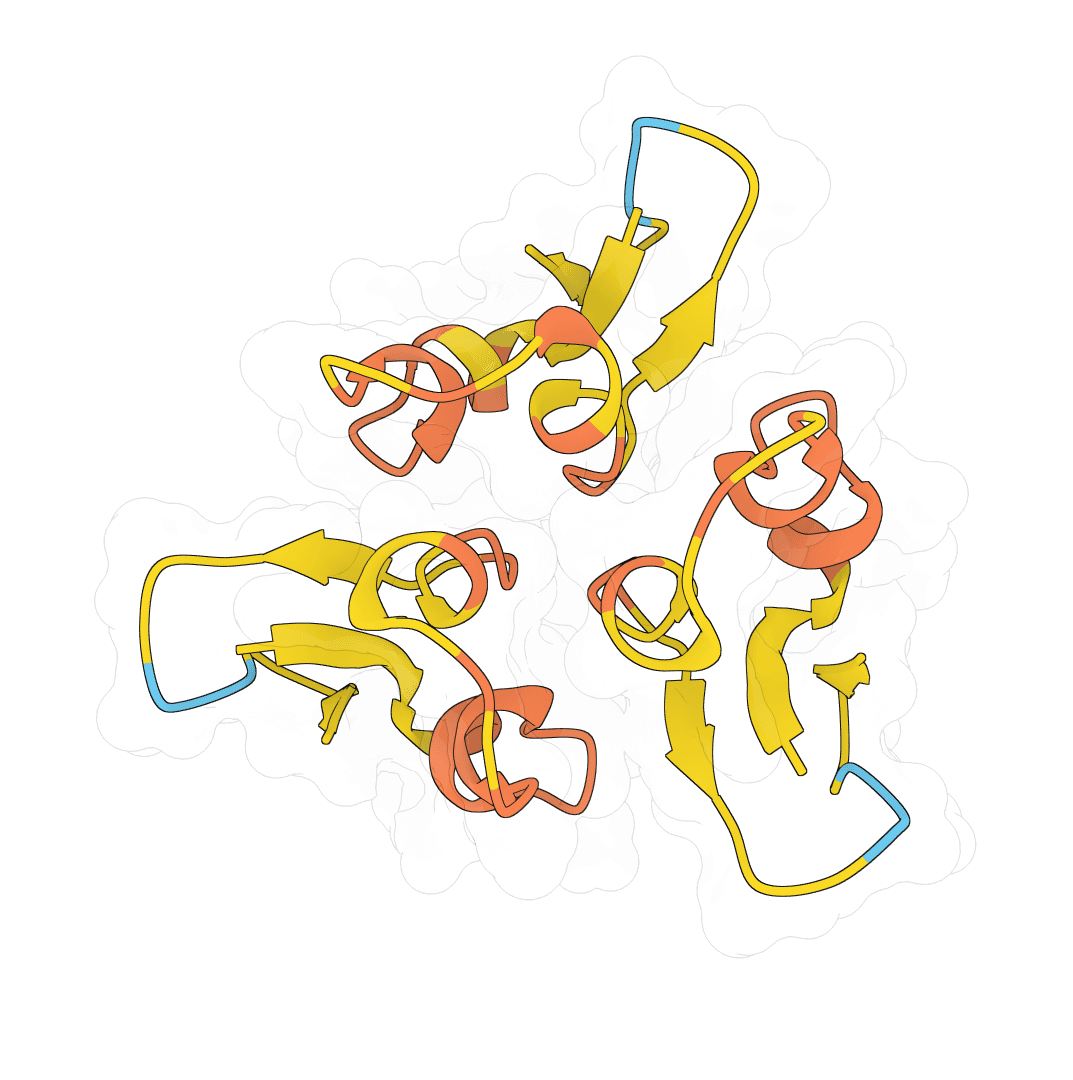
Rather than relying on a single model, ImmuneBuilder generates four independent structure predictions. The structures are aligned, and the one closest to the ensemble average is selected as the final prediction. Disagreement between ensemble members indicates uncertainty—high variance at a residue position signals low confidence in that region's conformation.
Raw neural network outputs occasionally contain steric clashes or stereochemical errors. ImmuneBuilder addresses this with a refinement step using OpenMM and the AMBER14 force field. Restrained energy minimization keeps backbone heavy atoms near their predicted positions while resolving physically implausible geometries.
ABodyBuilder2 was trained on 7,084 antibody structures from the Structural Antibody Database (SAbDab), filtered for resolution below 3.5 Angstroms with complete variable regions. Separate models were developed for nanobodies and TCRs using analogous curated datasets.
ProteinIQ hosts ImmuneBuilder 1.2 on GPU infrastructure, delivering structure predictions for immune receptors in under a minute without local installation.
| Input | Description |
|---|---|
Heavy/Alpha Chain | VH chain (antibody), VHH domain (nanobody), or alpha chain (TCR). FASTA or raw amino acid sequence. |
Light/Beta Chain | VL chain (antibody) or beta chain (TCR). Optional for nanobodies, which are single-domain. |
The tool auto-detects receptor type based on input chains:
For T-cell receptors, select TCRBuilder2 manually when you know the input is an alpha/beta receptor. In auto mode, ProteinIQ tries the native antibody model for two-chain inputs first and falls back to native TCRBuilder2 only if ANARCI rejects the antibody chain identity.
| Setting | Description |
|---|---|
Model type | Auto-detect treats single-chain inputs as nanobodies. For two-chain inputs, it tries ABodyBuilder2 first and falls back to TCRBuilder2 if ANARCI rejects antibody chain identity. Override with TCRBuilder2 for known alpha/beta T-cell receptor inputs. |
Numbering scheme | Controls the residue numbering written into the output structure. ImmuneBuilder supports imgt, chothia, kabat, aho, wolfguy, martin, and raw. |
TCR weights | When using TCRBuilder2, choose between the default TCRBuilder2+ weights and the original TCRBuilder2 weights for reproducibility. |
Include ensemble artifacts | Returns the refined prediction plus ranked unrefined models and the raw error_estimates.npy file produced by save_all(). |
Manual model selection is still preferred for TCRs, which use two chains but rely on a different published model family.
ImmuneBuilder outputs a PDB file containing the predicted 3D structure with per-residue error estimates stored in the B-factor column. Lower values indicate higher agreement across the ensemble. The interactive viewer displays the structure directly from this refined PDB.
| Metric | Description |
|---|---|
Mean predicted error | Mean residue-level B-factor across the refined structure. Lower values indicate more reliable predictions. |
Model used | Which ImmuneBuilder variant (ABodyBuilder2, NanoBodyBuilder2, TCRBuilder2, or TCRBuilder2+) generated the prediction. |
Total residues | Number of amino acids in the predicted structure. |
The reported error estimates derive from ensemble disagreement—residues where all four models predict similar conformations receive lower error values, while variable regions receive higher values. These scores particularly help identify:
ImmuneBuilder addresses several needs in antibody and immune receptor research:
CDR-H3 prediction remains challenging across all methods. This hypervariable loop adopts diverse conformations depending on antigen context, and both experimental and computational approaches show limitations. ImmuneBuilder's 2.81 Angstrom RMSD for CDR-H3 represents state-of-the-art performance but may not capture all relevant conformational states.
The models were trained on crystallographic structures, which represent static snapshots. Dynamic conformational changes upon antigen binding or other induced-fit mechanisms are not captured.
Performance may degrade for antibodies with unusual features not well-represented in training data, such as very long or short CDR loops, unusual disulfide patterns, or engineered modifications.