Design protein sequences with atomic context from ligands, metals, and nucleotides. Achieves 63.3% sequence recovery at binding sites, significantly outperforming ProteinMPNN (50.5%).
Design protein sequences for given backbone structures using deep learning. Fast and accurate inverse folding with state-of-the-art sequence recovery (52.4%).
Specialized model for soluble protein sequence design. Trained exclusively on soluble proteins for optimized performance on cytoplasmic and extracellular proteins.
Design antibody heavy- and light-chain CDR sequences from an antibody-antigen complex with the IgDesign inverse-folding model.
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
Inverse folding with ESM-IF1. Design protein sequences for given 3D backbone structures using a geometric deep learning model. Generate multiple sequence variants optimized for your target structure.

ProFam-1 is a protein family language model for family-conditioned sequence generation. Provide a protein family in FASTA, A2M, or A3M format and generate new sequences with model likelihood scores for downstream ranking and screening.
Design linear peptide binders for target proteins using a target sequence-conditioned masked language model. PepMLM generates peptide sequences optimized to bind specific protein targets based on ESM-2 protein language modeling.
Design de novo protein binders using AlphaFold2 backpropagation, ProteinMPNN sequence optimization, and PyRosetta relaxation. BindCraft generates novel protein sequences that bind to user-specified target surfaces.
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Design protein sequences with atomic context from ligands, metals, and nucleotides. Achieves 63.3% sequence recovery at binding sites, significantly outperforming ProteinMPNN (50.5%).
Design protein sequences for given backbone structures using deep learning. Fast and accurate inverse folding with state-of-the-art sequence recovery (52.4%).
Specialized model for soluble protein sequence design. Trained exclusively on soluble proteins for optimized performance on cytoplasmic and extracellular proteins.
Design antibody heavy- and light-chain CDR sequences from an antibody-antigen complex with the IgDesign inverse-folding model.
Inverse folding for antibody variable domains and nanobodies. Predicts amino acid sequences compatible with antibody structures using IMGT numbering while preserving native AntiFold chain handling and structural constraints.
Inverse folding with ESM-IF1. Design protein sequences for given 3D backbone structures using a geometric deep learning model. Generate multiple sequence variants optimized for your target structure.
ProFam-1 is a protein family language model for family-conditioned sequence generation. Provide a protein family in FASTA, A2M, or A3M format and generate new sequences with model likelihood scores for downstream ranking and screening.
Design linear peptide binders for target proteins using a target sequence-conditioned masked language model. PepMLM generates peptide sequences optimized to bind specific protein targets based on ESM-2 protein language modeling.
Design de novo protein binders using AlphaFold2 backpropagation, ProteinMPNN sequence optimization, and PyRosetta relaxation. BindCraft generates novel protein sequences that bind to user-specified target surfaces.
Score protein mutations with evolutionary profiles from homologous sequences and inverse folding. EvoIF returns a dimensionless log-odds score for each submitted single or multi-site mutation.
Configure inputs to begin
Set options on the left, then click “Submit job”.
Many thermostability design projects start with a solved or predicted protein structure and a specific question: which amino acid sequence is more likely to keep this backbone folded at high temperature? HyperMPNN answers that inverse-folding question by using ProteinMPNN weights retrained on proteins from hyperthermophilic organisms.
The native HyperMPNN repository is not a separate inference engine. It supplies retrained model weights that run through the original ProteinMPNN code path with --path_to_model_weights and --model_name. ProteinIQ follows that standard behavior and supports the HyperMPNN checkpoints as protein-only sequence design models.
HyperMPNN is most useful when thermal resilience is the design objective and the backbone is already chosen. Typical inputs include enzyme structures for high-temperature biocatalysis, vaccine nanoparticle components that need better storage stability, and scaffold proteins where functionally important residues can be fixed while the rest of the sequence is redesigned.
HyperMPNN runs online from one protein backbone, supplied as a PDB file or RCSB PDB ID, with a chosen number of thermostability-biased sequence variants. ProteinIQ returns designed sequences, mutation lists, native ProteinMPNN scores, sequence recovery, a FASTA file, and optional score or probability arrays for downstream analysis.
| Input | Accepted formats | Notes |
|---|---|---|
Protein | .pdb, .ent, or 4-character RCSB PDB ID such as 1CRN | The structure must contain protein atoms. HyperMPNN designs sequence for the provided backbone geometry and does not predict a new backbone. |
The input structure determines what HyperMPNN can preserve. Fixed catalytic residues, metal-binding residues, disulfide cysteines, interface hot spots, and experimentally required mutations should be constrained before running broad redesigns.
| Setting | Default | Description |
|---|---|---|
HyperMPNN model | v48_020_epoch300_hyper | Checkpoint from the native HyperMPNN retraining set. The default 0.20 A noise model matches the main native example and standard ProteinMPNN training noise. |
Number of sequences | 1 | Number of designs to sample, from 1 to 200. Initial screens usually benefit from 8 to 10 designs; the published HyperMPNN validation command sampled 200. |
Sampling temperature | 0.1 | Diversity control from 0.001 to 1.0. The published validation command used 0.001 for conservative sampling. Values around 0.1 to 0.3 explore progressively more substitutions. |
Random seed | 0 | ProteinMPNN treats 0 as random seed selection. Results report the effective nonzero seed selected for the run. A submitted nonzero seed makes the run reproducible with identical inputs and settings. |
| Setting | Description |
|---|---|
Chains to design | Comma-separated chain IDs such as A,B. Omitted chains stay fixed. Leaving the field empty designs all chains. |
Homo-oligomer | Ties equivalent positions across selected chains, so homomeric chains receive the same sequence. Selected chains must have matching residue counts. |
Fixed positions | Positions that must remain unchanged, using entries such as A15, A19, A1-10, or B. Fixed positions protect active sites, binding residues, and known stabilizing mutations. |
Redesigned positions | Positions allowed to change; all other positions are fixed. This is useful for loop redesigns, surface patches, and local thermostability experiments. |
Exclude amino acids | One-letter amino acid codes to omit globally, such as C or CW. Exclusions help when unwanted cysteines, oxidation-prone residues, or rare residues would complicate experiments. |
Save score file | Saves native score arrays as .npz files for custom ranking or offline analysis. |
Save probability file | Saves per-position probability arrays as .npz files for uncertainty analysis, sequence logos, or residue-level sampling diagnostics. |
Amino acid biases | Adds global sampling biases for individual amino acids. Positive values increase sampling frequency. Negative values reduce it. A value near -25 effectively excludes an amino acid. |
HyperMPNN returns both a human-readable table and native files. The FASTA output is the primary scientific output because it preserves ProteinMPNN headers and metadata.
| Output | Meaning |
|---|---|
Sequence | Designed amino acid sequence. Multi-chain designs are separated in the native FASTA style. |
Score | Average negative log probability over designed residues. Lower values mean the model assigned higher probability to the sampled sequence at the redesigned positions. |
Global score | Average negative log probability over all residues in the structure. Lower values are better for overall sequence fit to the backbone. |
Sequence recovery | Fraction of redesigned positions that match the input sequence. Low recovery means the model changed many residues; high recovery means the design stayed close to the starting sequence. |
Mutations | Substitutions relative to the input structure, reported with chain and residue context. |
Identity | Percent identity to the original sequence over the designed region. |
FASTA file | native ProteinMPNN sequence output with native and sampled records. |
Designed FASTA | Sampled designs only, with one standard FASTA record per designed chain for workflow reuse. |
Score NPZ | Optional native score arrays when Save score file is enabled. |
Probability NPZ | Optional per-position probability arrays when Save probability file is enabled. |
Provenance JSON | HyperMPNN and ProteinMPNN commits, checkpoint identity, submitted seed, and effective seed used for the run. |
For multichain designs, the Results view separates each sampled design by chain while the Data table keeps one row per complete sampled design. The primary FASTA file preserves ProteinMPNN's combined multichain records and headers; workflows use chain-specific records from the designed-only FASTA so the input sequence is not treated as another design.
Jobs are limited by total padded structure residues multiplied by requested sequences: up to 250,000 residue-samples normally or 100,000 when probability arrays are requested. Fixed context chains and PDB numbering gaps count because ProteinMPNN featurizes the complete structure. This keeps large sequence and probability outputs within the execution and result-transfer envelope.
HyperMPNN scores rank sequences by model likelihood, not by measured melting temperature. A low Score or Global score indicates that the sequence is compatible with the input backbone under the HyperMPNN model, but experimental thermostability still depends on expression, folding kinetics, oligomerization, cofactors, and the validity of the backbone.
Global score is the better broad ranking field for complete designs. Score is more useful when the redesign is local and fixed residues dominate the structure. When both scores are similar across candidates, mutation patterns usually matter more than small score differences.
Sequence recovery has a different meaning in thermostability redesign than in native-sequence recovery benchmarks. A low value is not automatically bad. HyperMPNN is expected to move sequences toward hyperthermophile-like composition, so useful designs may have substantial substitutions on the surface, in the core, or around flexible regions.
8 to 10 designs at Sampling temperature 0.1 or 0.15 gives enough diversity for a first pass.Global score, reasonable mutation load, and no obvious disruption of active-site chemistry or interface contacts.Fixed positions, followed by stability or function-specific evaluation of the redesigned structures.HyperMPNN fine-tunes the ProteinMPNN inverse-folding architecture on structures from hyperthermophilic organisms. The published workflow started from 96,738 sequences, clustered them at 50 percent identity, filtered AlphaFold2-predicted structures by pLDDT above 70, and trained on 29,042 protein structures from organisms adapted to extreme heat.
During inference, HyperMPNN receives the same information as ProteinMPNN: backbone geometry, chain selection, fixed-position masks, tied positions, amino acid omissions, and optional amino acid biases. The difference is the learned sequence distribution. HyperMPNN shifts sampling toward amino acid patterns observed in thermostable proteins rather than the broader Protein Data Bank distribution.
The HyperMPNN paper reports several composition-level trends in hyperthermophilic proteins and HyperMPNN designs:
| Structural region | Reported shift versus mesophilic proteins |
|---|---|
| Surface | More positively charged residues |
| Surface | More apolar residues |
| Surface | Fewer polar uncharged residues |
| Core | More apolar residues |
These shifts are consistent with common thermostability mechanisms, including tighter hydrophobic packing and stronger electrostatic networks. HyperMPNN should not be read as a salt-bridge maximizer. The reported median salt bridge count in native hyperthermophilic proteins was close to the mesophilic comparison set, while HyperMPNN designs recovered hyperthermophile-like salt bridge counts better than standard ProteinMPNN in the authors' analysis.
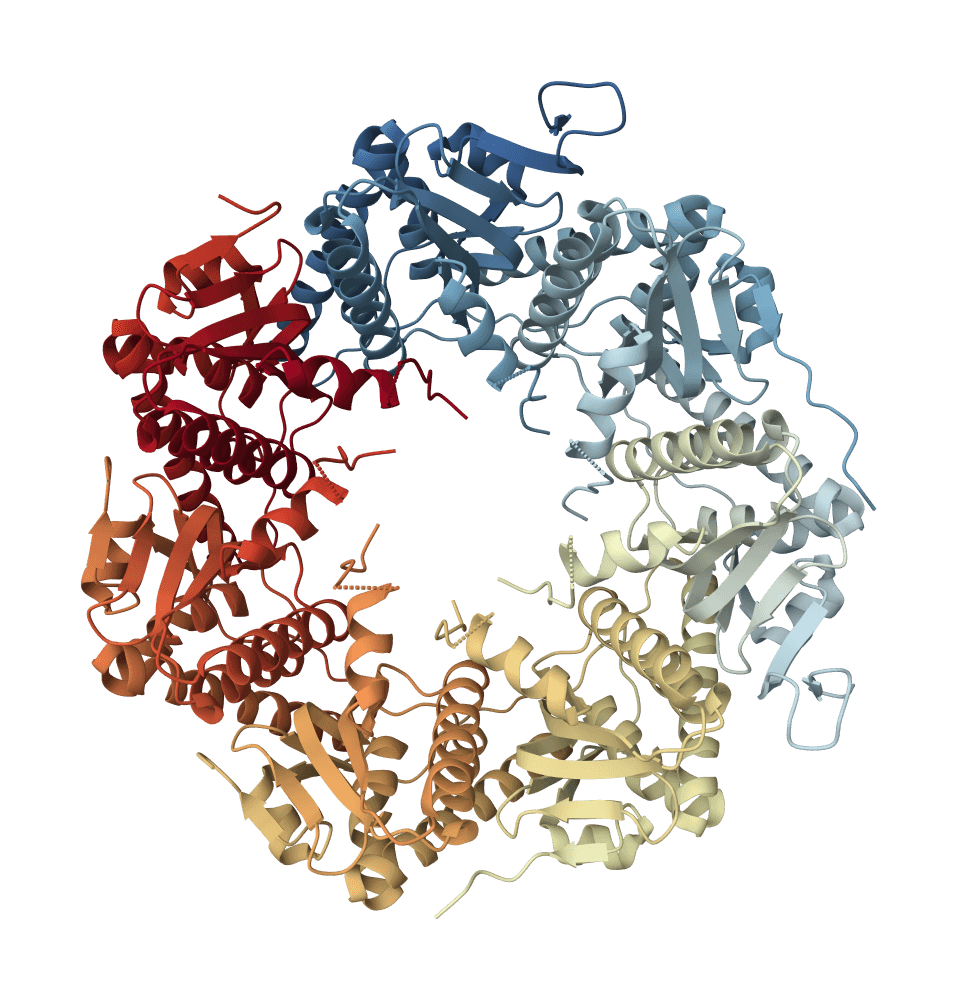
The experimental validation case in the HyperMPNN preprint used the I53-50B pentamer from an icosahedral nanoparticle system. Its published command sampled 200 sequences at temperature 0.001; the Published validation sampling preset reproduces that sampling envelope. Designs selected from the workflow retained stability at 95 degrees C, while the parent sequence had a melting temperature of 65 degrees C. That result is a strong proof of concept, not a guarantee that every redesigned backbone will gain 30 degrees C of thermal tolerance.
| Tool | Best fit | Main caveat |
|---|---|---|
| HyperMPNN | Full-sequence or region-specific redesign with thermostability as the main objective | Does not model ligands, cofactors, or assay-specific function directly. |
| ProteinMPNN | General inverse folding when no special property bias is desired | Less targeted toward hyperthermophile-like sequence composition. |
| LigandMPNN | Redesign around ligands, metals, nucleotides, or fixed side-chain context | Better for binding-site preservation than thermostability bias. |
| SolubleMPNN | Protein-only sequence design where soluble expression is the priority | Solubility and thermostability can conflict, so expression checks still matter. |
| ESM-IF1 | Alternative inverse-folding model for generating backbone-compatible sequences | Not specifically trained for thermostable sequence composition. |
| ThermoMPNN | Scoring point mutations for predicted stability changes | Mutation scoring does not generate full redesigned sequences. |
For structures that come from sequence rather than experiment, backbone prediction or refinement with AlphaFold2 should come before HyperMPNN. Low-confidence or flexible regions need cautious interpretation because HyperMPNN can only design against the geometry it receives.